Volume 6 (2023) Issue 4 No.5 Pages 135-138
Abstract
This article reports the clinical course and imaging findings of three cases of suspected pleuroparenchymal fibroelastosis (PPFE) after allogeneic hematopoietic cell transplantation (HCT). All patients complained of dyspnea more than 5 years after HCT, had progressive restrictive deficits on respiratory function tests, and presented with pneumothorax, pleural thickening, or exacerbation of consolidation in the upper lobe of the lung. Though lung biopsy was not done in all three cases, the clinical findings and results of spirometry were compatible with those of PPFE. PPFE has been sporadically reported as a pulmonary complication of allogeneic HCT; however, clinical diagnostic criteria other than histological diagnosis and treatment methods have not yet been established. The accumulation of more cases is necessary to improve the prognosis of PPFE complications.
Introduction
Pulmonary complications that develop after allogeneic hematopoietic cell transplantation (HCT) occur in one-third of the patients and have high mortality rates1. Bronchiolitis obliterans and cryptogenic organizing pneumonia are known post-transplant pulmonary complications associated with autoimmunity and graft-versus-host disease (GVHD). Pleuroparenchymal fibroelastosis (PPFE) is a restrictive pulmonary disease that causes fibrosis of the pleura and adjacent pulmonary parenchyma, predominantly in the upper lobe. The term “PPFE” was first used by Frankel et al. in 20042, and the idiopathic form is now listed as one of the rare idiopathic interstitial pneumonias in the classifications of the American Thoracic Society and the European Respiratory Society3. It progresses slowly but steadily, and the 5-year survival rate is < 30%, indicating a poor prognosis4. Recently, there have been sporadic reports on the development of PPFE after allogeneic HCT5; however, there are few comprehensive reports. The diagnosis is made based on characteristic imaging findings and histopathology, and clinical diagnostic criteria have not yet been established.
We encountered three patients with clinically suspected PPFE following allogeneic HCT. Here, we describe the clinical aspects and outcomes of each case and discuss an effective index for clinical diagnosis and risk factors for developing PPFE after allogeneic HCT.
Case Presentation
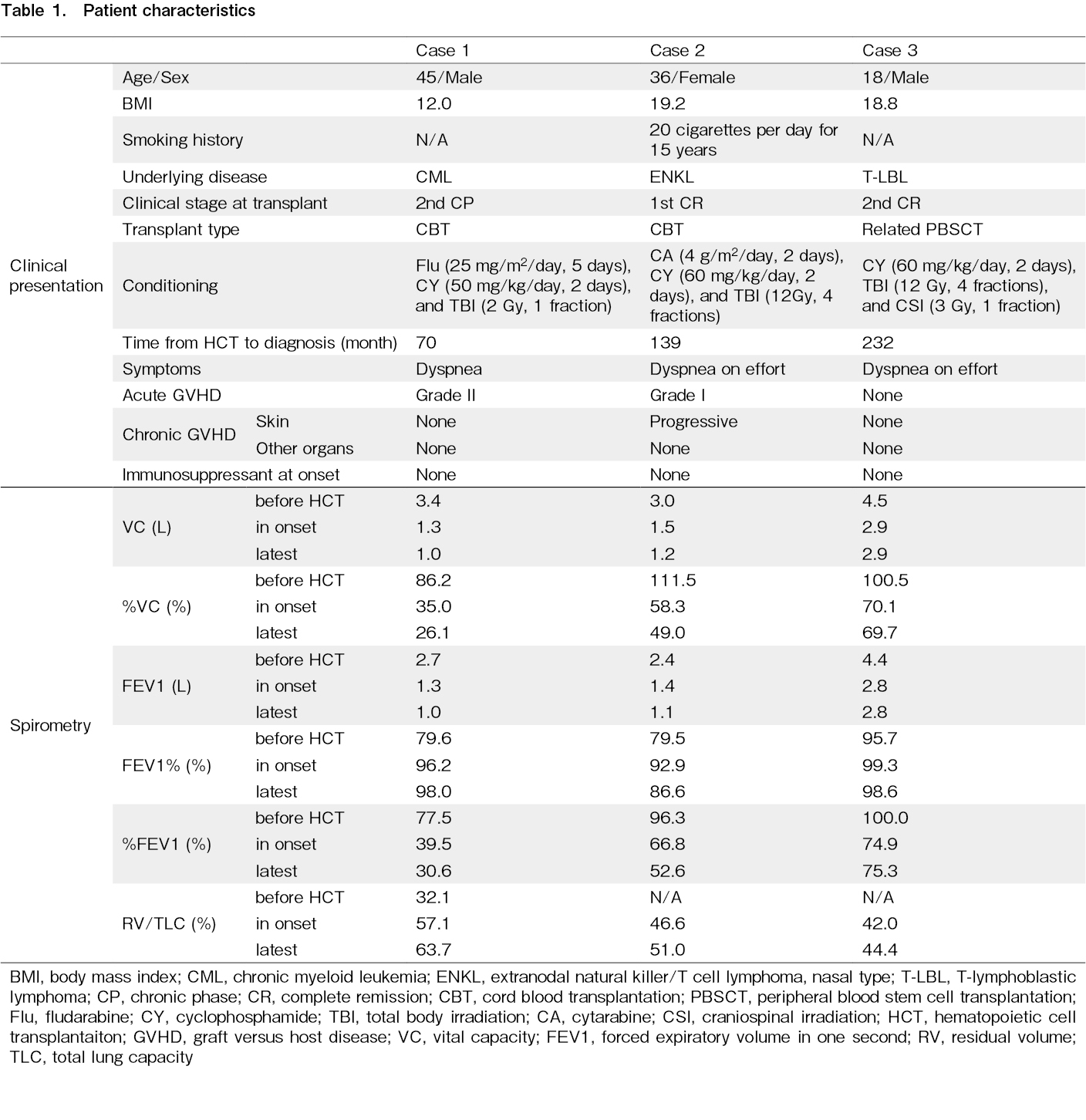
Patients' characteristics are listed in Table 1.
Case 1
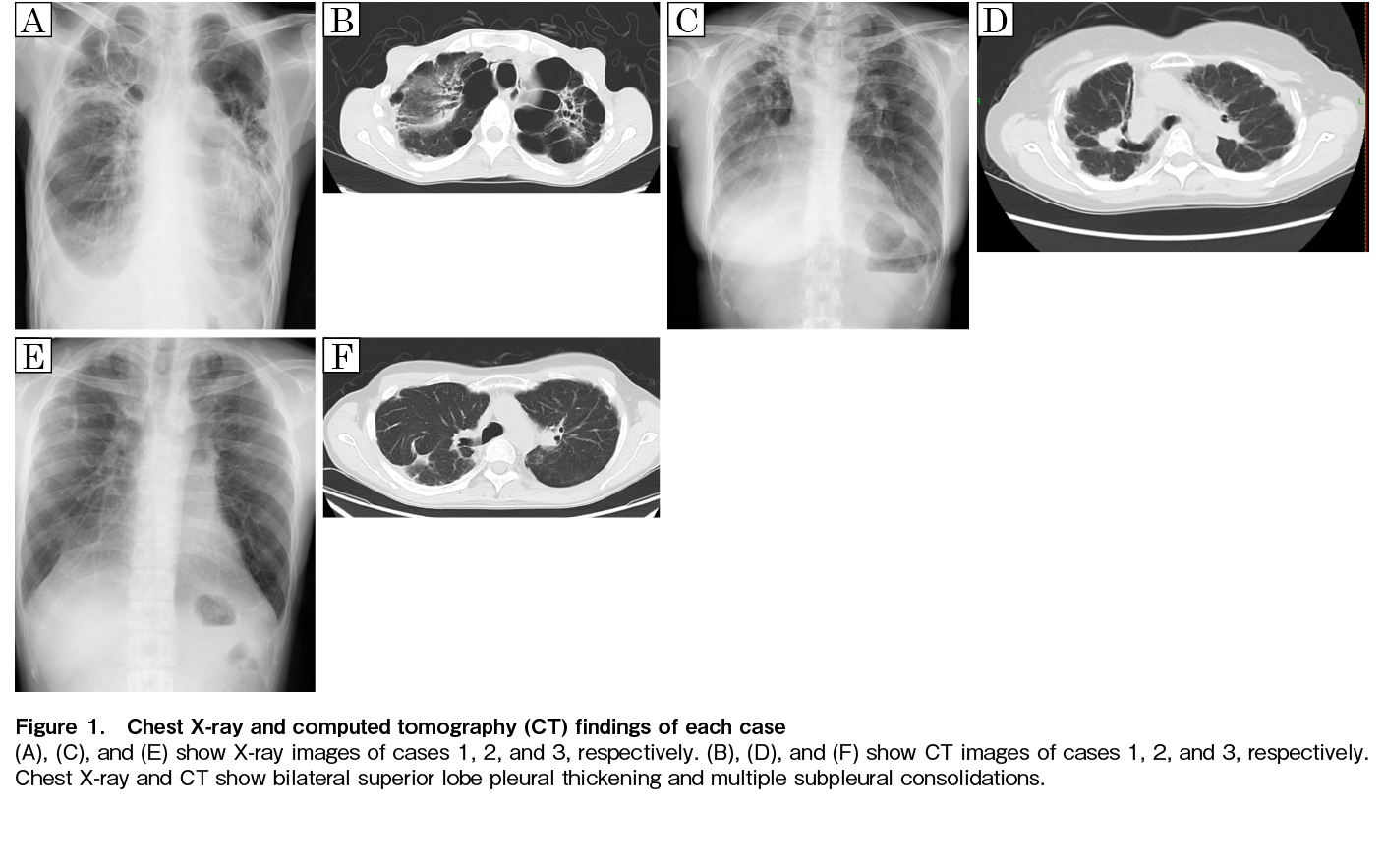
A 44-year-old man was diagnosed with a blast crisis in chronic myelogenous leukemia and underwent cord blood transplantation (CBT) at 45 years of age in the second chronic phase with the conditioning regimen of fludarabine (25 mg/m2/day, 5 days), cyclophosphamide (CY) (50 mg/kg/day, 2 days), and 2 Gy of total body irradiation (TBI). Before undergoing CBT, multiple bullae were observed in the bilateral upper lobes of the lungs. Six months after the transplantation, the patient developed a pneumothorax and was treated with thoracic drainage. Six years after the transplantation, symptoms such as cough and dyspnea gradually worsened, and chest X-ray revealed worsening opacities in the upper lung fields. Computed tomography (CT) revealed emphysematous bullae clusters in both lung apices and consolidation in the left upper lobe (Figure 1A and B). His restrictive disorder had progressed (FEV1.0 1.0L, FEV1.0% 98.0%, %VC 26.1%, RV/TLC 63.7%). Seven years after the transplantation, the patient required home oxygen therapy.
Case 2
A 35-year-old woman was diagnosed with extranodal NK/T-cell lymphoma that was suspected to be treatment-resistant. The patient underwent CBT at 36 years of age with the conditioning regimen of cytarabine (4 g/m2/day, 2 days), CY (60 mg/kg/day, 2 days), and 12 Gy of TBI. Five years after the transplantation, chest X-ray revealed suspected pleural thickening in the right apex, and CT showed bilateral pleural thickening and wedge-shaped consolidation (Figure 1C and D). Eleven years after the transplantation, she had a slow decline in respiratory function (FEV1.0 1.1L, FEV1.0% 86.6%, %VC 49.0%, RV/TLC 51.0%), but no demand for oxygen.
Case 3
An 18-year-old man in second remission of T-lymphoblastic lymphoma underwent allogeneic peripheral blood stem cell transplantation from an HLA-matched sibling donor with the conditioning regimen of CY (60 mg/kg/day, 2 days), 12 Gy of TBI, and 3 Gy of craniospinal irradiation. Nine years after the transplantation, dyspnea on exertion and restrictive disability progressed. Chest X-ray revealed mild pleural thickening at the apex of the lung, and CT showed wedge-shaped consolidation just below the pleura of both the upper lobes and flattening of the thorax (Figure 1E and F). Although gradual progression of restrictive disability was observed (FEV1.0 2.8L, FEV1.0% 98.6%, %VC 69.7%, RV/TLC 44.4%), periodic respiratory function tests and follow-up were performed.
Discussion
Advances in allogeneic HCT technology and an increase in long-term survivors have led to previously unknown late pulmonary complications after allogeneic HCT, the progression of which affects the quality of life of long-term survivors after allogeneic HCT. Therefore, accurate diagnosis and timely therapeutic intervention are desired. PPFE after allogeneic HCT has gained increasing recognition in recent years after a case report first published in 20116. The diagnosis is made based on radiographic imaging findings, including consolidation predominantly in the upper lung field and volume reduction in the upper lobe, and pathological findings, including fibrosis with poor inflammatory cell infiltration in the alveolar septum just below the pleura. Though there are many cases where it is difficult to perform a lung biopsy depending on the patient's medical condition, the international clinical diagnostic criteria that do not require a surgical lung biopsy have not yet been established. Watanabe et al. set RV/TLC
The risk of developing PPFE after transplantation is thought to be associated with the use of alkylating agents, irradiation, and respiratory infections; however, since these conditions can equally affect many allograft patients who have not had PPFE, they are not PPFE-specific5. The development of PPFE may be more complicated by the patients' backgrounds or other factors under transplant-related conditions. Therefore, we suggest that continuous pulmonary function testing may be important for all patients.
No drugs have been confirmed to prevent the progression of PPFE, and corticosteroids, immunosuppressive drugs, and antifibrotic agents have been used in some cases, with poor efficacy5, 9, 10. Since the symptoms progress slowly, the patients are often observed without treatment. Lung transplantation is the most effective treatment5, 9 but not an easy treatment approach. We did not administer any drugs to our three cases, and none of them received lung transplantation.
One of the reasons why the development of treatment methods for PPFE has not progressed is that the number of cases is small and it is difficult to organize clinical trials to verify therapeutic effects. Based on this experience, it is necessary to first establish diagnostic criteria that do not require histopathological diagnosis and to accumulate cases in the future to improve the prognosis of PPFE after HCT.
Author Contributions
SM collected and interpreted the data and wrote the manuscript. AK treated the patients. TA, TF, SN, YK, MG, NF, KO, and AK supervised the writing of the manuscript. All authors participated in the interpretation of the results and approved the final manuscript.
Informed Consent
Informed consent was obtained by all participants in this study.
Conflicts of Interest
The authors declare no conflict of interest. Disclosure forms provided by the authors are available on the website.
References
1.Haider S, Durairajan N, Soubani AO. Noninfectious pulmonary complications of haematopoietic stem cell transplantation. Eur Respir Rev. 2020; 29: 190119.
2.Frankel SK, Cool CD, Lynch DA, Brown KK. Idiopathic pleuroparenchymal fibroelastosis: description of a novel clinicopathologic entity. Chest. 2004; 126: 2007-13.
3.Travis WD, Costabel U, Hansell DM, King TE, Jr., Lynch DA, Nicholson AG, et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013; 188: 733-48.
4.Enomoto Y, Nakamura Y, Satake Y, Sumikawa H, Johkoh T, Colby TV, et al. Clinical diagnosis of idiopathic pleuroparenchymal fibroelastosis: A retrospective multicenter study. Respir Med. 2017; 133: 1-5.
5.Arndt P. Pleuroparenchymal Fibroelastosis: A Review with a Focus on a Non-Infectious Complications after Hematopoietic Stem Cell Transplant. Biomedicines. 2023; 11: 924.
6.von der Thusen JH, Hansell DM, Tominaga M, Veys PA, Ashworth MT, Owens CM, et al. Pleuroparenchymal fibroelastosis in patients with pulmonary disease secondary to bone marrow transplantation. Mod Pathol. 2011; 24: 1633-9.
7.Watanabe K, Ishii H, Kiyomi F, Terasaki Y, Hebisawa A, Kawabata Y, et al. Criteria for the diagnosis of idiopathic pleuroparenchymal fibroelastosis: A proposal. Respir Investig. 2019; 57: 312-20.
8.Ishii H, Kinoshita Y, Kushima H, Nagata N, Watanabe K. The similarities and differences between pleuroparenchymal fibroelastosis and idiopathic pulmonary fibrosis. Chron Respir Dis. 2019; 16: 1479973119867945.
9.Chua F, Desai SR, Nicholson AG, Devaraj A, Renzoni E, Rice A, et al. Pleuroparenchymal Fibroelastosis. A Review of Clinical, Radiological, and Pathological Characteristics. Ann Am Thorac Soc. 2019; 16: 1351-9.
10.Bondeelle L, Gras J, Michonneau D, Houdouin V, Hermet E, Blin N, et al. Pleuroparenchymal fibroelastosis after allogeneic hematopoietic stem cell transplantation. Bone Marrow Transplant. 2020; 55: 982-6.
Search
News


