Volume 8 (2025) Issue 1 No.8 Pages 186-189
Abstract
Background: Allogeneic hematopoietic cell transplantation (Allo-HCT) is the only curative option for marrow failure, myelodysplastic syndrome (MDS), and acute myeloid leukemia associated with dyskeratosis congenita (DKC). Due to chromosomal instability and sensitivity to radiation and alkylating agents, HCT is associated with a high incidence of transplant-related mortality in DKC.
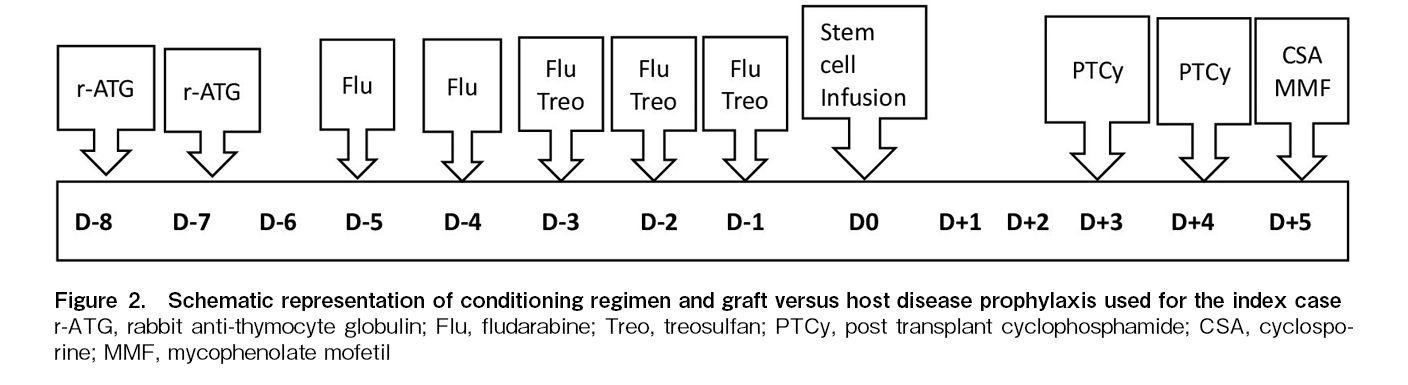
Case report: A 25-year-old male presented with DKC-associated cutaneous manifestations and myelodysplastic syndrome / acute myelogenous leukemia (MDS/AML). Targeted next-generation sequencing revealed mutation of the DKC1 and RUNX1 genes. His mother and sibling sisters were carriers for the DKC1 mutation. Due to high donor-specific antibody mean fluorescence intensity (DSA-MFI) against the unshared Human Leukocyte Antigen-A (HLA-A) allele of his 6/12 HLA-matched father, his paternal cousin's sister was selected as a haploidentical (6/12 HLA-matched) donor for HCT. He underwent allo-HCT with stable disease burden using a specifically-designed RIC regimen containing treosulfan (at 50% reduced dosing), fludarabine, and rabbit anti-thymocyte globulin. The graft versus host disease (GVHD) prophylaxis contained reduced-dose post-transplant cyclophosphamide (PTCy dose reduction of 50%) with mycophenolate mofetil and cyclosporine. He engrafted with complete donor chimerism, and the day +30 marrow was in complete morphological remission with undetectable measurable residual disease by flow cytometry. On day +126, he developed steroid-responsive late-onset grade II acute GVHD (stage III skin GVHD). He suffered from morphologic relapse on day +220 and succumbed from sepsis with septic shock on day +256.
Conclusion: This case demonstrates the safety and feasibility of haploidentical-HCT using a treosulfan-based reduced-intensity conditioning (RIC) regimen and modified PTCy-based GVHD prophylaxis in DKC. Disease relapse in this patient underscores the impact of pretransplant disease burden on relapse free survival in DKC patients with MDS/AML who are not eligible for myeloablative conditioning.
Introduction
Dyskeratosis congenita (DKC) is a telomeropathy resulting from mutations in genes involved in the maintenance of telomeres, including DKC1, TERT, TERC, NOP10, NHP2, TCAB1, RTEL1, and TINF21. The classic triad of oral leukoplakia, skin hyperpigmentation, and dystrophic nails is common with DKC mutation, though these features may be subtle or present with varying combinations2. Allogenic hematopoietic stem cell transplantation (Allo-HCT) is the only curative therapy of severe bone marrow failure (BMF), myelodysplastic syndrome (MDS), and acute myeloid leukemia (AML) in DKC patients, though it does not correct other organ dysfunctions associated with telomeropathy, such as liver fibrosis and pulmonary fibrosis1. Historically, long-term outcomes after myeloablative HCT have been poor in DKC patients3. In this case report, we have described the feasibility of haploidentical HCT using a rationally designed reduced intensity conditioning (RIC) regimen with a modified graft versus host disease (GVHD) prophylaxis protocol in a DKC patient with MDS/AML.
Case Description
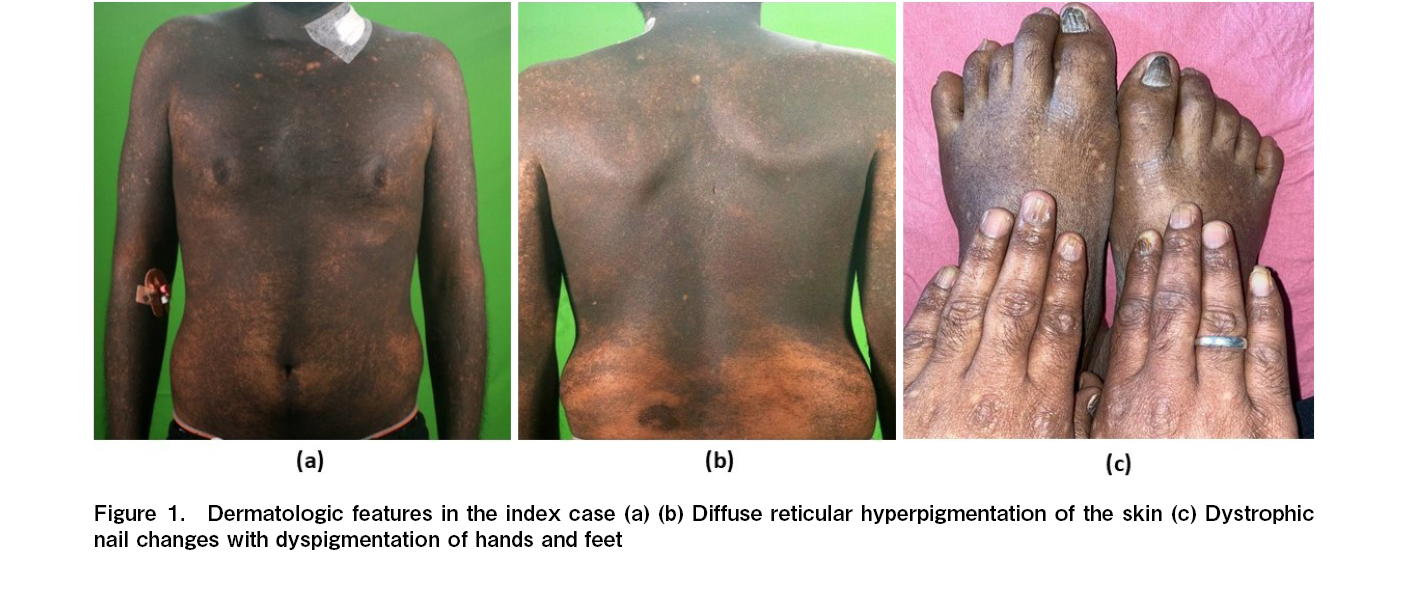
A 25-year-old male with generalized hyperpigmented skin lesions and dystrophic nails presented to our center with progressive pancytopenia for one year. Detailed history revealed insidious onset hyperpigmented skin lesions from 14 years of age, which gradually increased in extent to involve 100% of body surface areas at 18 years of age. Clinical examination showed hyperpigmented reticular skin lesions (Figure 1A and 1B) and dystrophic nails (Figure 1C). A clinical diagnosis of DKC with BMF was suspected. Bone marrow aspiration with trephine biopsy showed 14% blasts, and flow cytometry of marrow aspirate was consistent with clonal myeloid blasts. Targeted next-generation sequencing for bone marrow failure syndrome panel genes performed on bone marrow aspirate samples revealed a likely pathogenic missense mutation in the DKC1 gene (X-linked recessive) at exon 12 (c.1223C>T) with a variant allele frequency (VAF) of 95% and a pathogenic RUNX1 gene at exon 3 (c.297C>A) with a VAF of 19%. A conventional karyotype performed on bone marrow aspirate samples was normal (46XY). Sanger sequencing in peripheral blood samples of the mother and both sibling sisters detected the same DKC1 mutation in heterozygous condition. A diagnosis of DKC with MDS/AML was made based on clinical findings, molecular genetics, and the presence of carrier states for the same DKC1 mutation in the mother and sisters. An ultrasound of the abdomen and a high-resolution computed tomography (HRCT) scan of the chest did not show any hepatic or pulmonary comorbidity. He had no prior history of malignancy. He was started on single agent Azacytidine 75 mg/m2, once daily for 7 days in each cycle. With each cycle of azacytidine he developed febrile neutropenia with recurrent soft tissue infection by methicillin-sensitive staphylococcus aureus (MSSA). After 6 cycles of single agent azacytidine, he had stable disease with 15% blasts in marrow aspirate. HLA typing of the parents and both sibling sisters were done, and all were 6/12 HLA-matched with the index case. The donor-specific antibody (DSA) mean fluorescence intensity (MFI) was 15,000 against the unshared HLA-A allele of the father. In extended family HLA typing on the paternal side, there was one 6/12 HLA-matched unmarried cousin's sister. The DSA MFI was 1,782 against the unshared HLA-B allele and 1,656 against the HLA-C allele. The cousin's sister was chosen as the prospective HCT donor. His age-adjusted HCT-CI score was
Discussion
Allogeneic hematopoietic cell transplantation in DKC patients is challenging because of the inherent sensitivity to chemotherapy and radiotherapy resulting in increased early and late toxicities and the frequent presence of pulmonary and hepatic comorbidities. Late death after HCT in this patient population results from gastrointestinal and pulmonary complications, as well as development of second cancers4,5.
To overcome these challenges, we specifically designed an RIC regimen containing rabbit ATG, fludarabine and reduced-dose treosulfan. The GVHD prophylaxis regimen was modified by a 50% dose reduction of PTCy.
This transplant presented specific challenges. Firstly, there was a high risk of early and late toxicity, which may have resulted from the conditioning regimen and PTCy. Secondly, the disease was not in remission before transplant, which requires a conditioning regimen with a strong anti-myeloid property. Thirdly, it was necessary to overcome the immunological barrier of haploidentical-HCT in a polytransfused patient with a high risk of alloimmunization to ensure engraftment and continued graft function. We choose treosulfan, an alkylating agent with strong efficacy against myeloid cancer6 albeit with lesser toxicity7. To make the RIC regimen, we empirically decreased the myeloablative dose of treosulfan by 50%. Fludarabine was included primarily as an immunosuppressive agent in the context of haplo-HCT. The timing of ATG administration is also important. The mean half-life of rabbit-ATG is close to 30 days8. Compared to early administration (before day -5), late administration (close to day 0) of ATG likely results in a greater degree of T-cell depletion in the graft and is expected to result in more viral infections9 and a higher incidence of relapse10. The use of rabbit ATG on days -8 and -7 in our protocol had a dual purpose of immunosuppression to prevent graft rejection as well as GVHD.
Previous reports using an RIC regimen in DKC have shown varying rates of primary as well as secondary graft failure and levels of mixed chimerism3,11. In contrast, a study by Nelson et al. reported complete donor chimerism and no early or late graft failure in a cohort of 7 patients1. Our regimen achieved complete donor chimerism with a well-functioning graft. Use of fludarabine and ATG in our conditioning regimen provided the needed potent immunosuppression to facilitate complete donor cell engraftment.
Similarly, to reduce the complications of PTCy, we reduced the dose of this alkylating agent by 50%. The absence of grade III-IV acute GVHD and chronic GVHD in our case shows the efficacy and safety of this modified GVHD prophylaxis regimen in DKC patients. Studies have shown the efficacy of low-dose PTCy/ATG for GVHD prophylaxis in haploidentical peripheral blood stem cell transplants. In this protocol, ATG was given on days -2 to -1 and a single dose of PTCy 50 mg/kg was administered on day +312.
The relapse of AML on day +226 in this patient is not unusual as the HCT was performed using an RIC regimen in high-risk MDS/AML (RUNX1-mutated in this case) and the disease was not in morphological remission at pretransplant.
One limitation of this case report is that germline testing for the DKC1 variant was not done. This missense variant is likely pathogenic as it results in the amino acid substitution of isoleucine for threonine at codon 408 and has been reported previously as a novel mutation in DKC showing genotype-phenotype correlation13. The high VAF with genotype-phenotype correlation and presence of a carrier state in the mother and siblings, strongly suggest the germline nature of the mutation.
This case demonstrates the feasibility and safety of this novel conditioning regimen with a modified GVHD prophylaxis protocol in this challenging and vulnerable group of patients. The relapse of the AML in this patient underscores the impact of disease control before transplant, especially in a population of patients who are at very high risk of morbidity and mortality from myeloablative transplant.
Author's Contribution
SSR, MM, and AK drafted the original manuscript. SSR and AK contributed to the concept and design of the report; MM provided the relevant images. SSR, MM, AK, PR, AJ, GP, and PM contributed to patient care. All authors critically reviewed and approved the final version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. Disclosure forms provided by the authors are available on the website.
Informed Consent
Written informed consent was taken from the patient.
References
1.Nelson AS, Marsh RA, Myers KC, Davies SM, Jodele S, O'Brien TA, et al. A Reduced-Intensity Conditioning Regimen for Patients with Dyskeratosis Congenita Undergoing Hematopoietic Stem Cell Transplantation. Biol Blood Marrow Transplant. 2016; 22: 884-8.
2.Womer R, Clark JE, Wood P, Sabio H, Kelly TE. Dyskeratosis congenita: two examples of this multisystem disorder. Pediatrics. 1983; 71: 603-9.
3.Gadalla SM, Sales-Bonfim C, Carreras J, Alter BP, Antin JH, Ayas M, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with dyskeratosis congenita. Biol Blood Marrow Transplant. 2013; 19: 1238-43.
4.Amarasinghe K, Dalley C, Dokal I, Laurie A, Gupta V, Marsh J. Late death after unrelated-BMT for dyskeratosis congenita following conditioning with alemtuzumab, fludarabine and melphalan. Bone Marrow Transplant. 2007; 40: 913-4.
5.Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009; 113: 6549-57.
6.Sjöö F, Hassan Z, Abedi-Valugerdi M, Griskevicius L, Nilsson C, Remberger M, et al. Myeloablative and immunosuppressive properties of treosulfan in mice. Exp Hematol. 2006; 34: 115-21.
7.Zhu S, Liu G, Liu J, Chen Q, Wang Z. Long-Term Outcomes of Treosulfan- vs. Busulfan-Based Conditioning Regimen for Patients with Myelodysplastic Syndrome and Acute Myeloid Leukemia Before Hematopoietic Cell Transplantation: A Systematic Review and Meta-Analysis. Front Oncol. 2020; 10: 591363.
8.Bunn D, Lea CK, Bevan DJ, Higgins RM, Hendry BM. The pharmacokinetics of anti-thymocyte globulin (ATG) following intravenous infusion in man. Clin Nephrol. 1996; 45: 29-32.
9.Lindemans CA, Chiesa R, Amrolia PJ, Rao K, Nikolajeva O, de Wildt A, et al. Impact of thymoglobulin prior to pediatric unrelated umbilical cord blood transplantation on immune reconstitution and clinical outcome. Blood. 2014; 123: 126-32.
10.Admiraal R, Jol-van der Zijde CM, van Kesteren C, Knibbe CAJ, Bredius R, Boelens JJ, et al. Exposure of thymoglobulin is associated with overall survival in children receiving allogeneic HCT: Towards individualized dosing to improve survival. Biol Blood Marrow Transplant. 2014; 20 (Suppl 1): S76-8.
11.Dietz AC, Orchard PJ, Baker KS, Giller RH, Savage SA, Alter BP, et al. Disease-specific hematopoietic cell transplantation: nonmyeloablative conditioning regimen for dyskeratosis congenita. Bone Marrow Transplant. 2011; 46: 98-104.
12.Xu X, Yang J, Cai Y, Li S, Niu J, Zhou K, et al. Low dose anti-thymocyte globulin with low dose posttransplant cyclophosphamide (low dose ATG/PTCy) can reduce the risk of graft-versus-host disease as compared with standard-dose anti-thymocyte globulin in haploidentical peripheral hematopoietic stem cell transplantation combined with unrelated cord blood. Bone Marrow Transplant. 2021; 56: 705-8.
13.Vulliamy TJ, Marrone A, Knight SW, Walne A, Mason PJ, Dokal I. Mutations in dyskeratosis congenita: their impact on telomere length and the diversity of clinical presentation. Blood. 2006; 107: 2680-5.
Search
News


