Volume 6 (2023) Issue 1 No.5 Pages 23-29
Abstract
Hematopoietic cell transplantation (HCT) is a potentially curative therapy for patients with high-risk malignant and nonmalignant conditions. Nevertheless, various post-allogeneic HCT (allo-HCT) complications with diverse chronology, etiology, and pathophysiological background can develop, including general and organ-specific complications, such as graft dysfunction, infectious, and non-infectious etiologies, as well as non-infectious pulmonary complications (NIPCs). Post-transplant complications can also be related to conditioning intensity and drug-specific side effects. However, treatment options for these complications are suboptimal at present.
Poor graft function (PGF) is a potentially life-threatening post-allo-HCT complication and is reported in 5-30% of patients. Nevertheless, consensus guidelines to define and treat PGF are not available. Most therapies are symptomatic with variable success rates. NIPCs are diverse and difficult to diagnose. The pathophysiology of NIPCs remains ill-defined, and effective treatment approaches have not been standardized, with mortality exceeding 50% for some conditions, such as idiopathic pneumonia syndrome (IPS). Modification of the conditioning regimen intensity and introduction of novel agents have been used to decrease post-allo-HCT complications, including infections, non-infectious complications, graft-versus-host disease (GvHD), as well as cardiopulmonary, neurological, hepatorenal, and other complications. Transplant-associated thrombotic microangiopathy (TA-TMA) is a lethal post-allo-HCT complication that may be associated with functional and genetic abnormalities in complement activation and related to the use of calcineurin inhibitors, such as cyclosporine and tacrolimus. The introduction of complement inhibitors has transformed TA-TMA from a lethal complication to a treatable syndrome.
Introduction
Hematopoietic cell transplantation (HCT) is a potentially curative therapy that has been widely performed for various genetic and acquired high-risk malignant and non-malignant diseases. However, highly intensified conditioning regimens can lead to serious morbidity and mortality, such as major organ dysfunction, life-threatening graft dysfunction, infections, bleeding, thrombosis, and non-infectious complications. Graft-versus-host disease (GvHD) is a major complication of allogeneic HCT (allo-HCT) procedures and constitutes a major additional cause of transplant-related morbidity and mortality; however, non-immune post-transplant complications are not uncommon.
Post-transplant complications can be organ specific and involve every organ in the body, with specific features that depend on the affected system. In this review, we address post-transplant complications, including Poor graft function (PGF), pulmonary complications, conditioning regimen modifications, and transplant-related thrombotic microangiopathy.
Defining and Treating Poor Graft Function after Hematopoietic Cell Transplants
PGF is an important and potentially life-threatening complication of allo-HCT. No consensus guidelines for defining PGF are available. Some authors as well as our team have defined PGF as a failure to recover granulocytes and platelets within 28 days post-transplant in patients with complete donor hematopoietic chimerism1. Patients with incomplete donor hematopoietic chimerism were excluded from the study. PGF is distinguished from graft rejection, a term that should only be used in the presence of convincing evidence to confirm recipient anti-donor immune response and graft failure, when the recipient recovers normal blood granulocyte and platelet counts but subsequently fails to meet the normal range for one or both2. Additionally, we excluded the concept that the bone marrow is the target of acute or chronic GvHD3 from our definition of PGF as we consider it incorrect. Data from animal models in which confounding effects of infection and anti-GvHD therapies can be excluded have provided no convincing support for this concept4.
Owing to these complexities in defining PGF and the different thresholds for defining normal blood granulocyte and platelet numbers, the reported incidence of PGF varies substantially. Reports indicate a range of 5-30%, depending on whether platelet count < 100 × 10E+9/L is included in the definition of PGF1–5.
Variables reported to be associated with the risk of PGF include auto-versus allotransplants, HLA- and ABO-disparities between donor and recipient, low-intensity pre-transplant conditioning regimens, low graft dose of nucleated bone marrow cells or CD34-positive cells, graft-type (blood versus bone marrow versus umbilical cord blood), fresh versus frozen grafts, intensive post-transplant immune suppression with bone marrow suppressing drugs, anti-donor human leukocyte antigen (HLA) antibodies, cytomegalovirus (CMV) and other infections and their therapies, iron overload, splenomegaly (mistakenly), acute and/or chronic GvHD (discussed above), GvHD therapy, and other factors1–5.
PGF can arise from abnormalities in hematopoietic stem or progenitor cell function, abnormalities in the bone marrow microenvironment, or abnormal interactions between them. Some data suggest that high levels of intracellular reactive oxygen species6,7; bone marrow microenvironmental abnormalities, including dysfunctional endothelial cells and abnormal mesenchymal stromal cells6,7; disease-related bone marrow abnormalities, such as myelofibrosis; and production of pro-inflammatory cytokines such as interleukin 2 (IL-2) and tumor necrosis factor-α (TNF-α), may also contribute to PGF8. Adverse effects of synchronous and/or metachronous conditions and their therapy, including infections, drugs, ionizing radiation GvHD, can cause or complicate PGF and hinder differential diagnosis.
Notably, no consensus guidelines for PGF treatment are available. Therapies include red blood cell (RBC) and platelet transfusions, intravenous immunoglobulin, molecularly cloned myeloid hematopoietic growth factors (granulocyte- and granulocyte/macrophage colony-stimulating factors [G- and G/M-CSF]), boost of donor blood or bone marrow cells, mesenchymal stromal cells, and/or a second transplant from the same or a different donor6,7. However, these interventions are only partially effective. Providing an additional boost of donor hematopoietic cells, including CD34-positive cells without the donor CD3 component, has variable efficacy9. G-CSF and GM-CSF sometimes improve blood granulocyte counts; however, these responses are short-lived. Second transplants are sometimes effective8. Evidence favoring the use of eltrombopag for PGF is based on retrospective studies and case reports10,11. A recent review of eltrombopag and romiplostim in post-transplant thrombocytopenia reported response in 70-80% recipients, most of which were transient12. Statins have been reported to improve in vitro bone marrow endothelial progenitor cell (EPC) function obtained from persons with PGF13. Two prospective clinical trials reported that N-acetyl-cysteine facilitated bone marrow recovery in persons with PGF; however, these findings are not convincing and need to be confirmed in future studies.
In conclusion, there is considerable disagreement and confusion in defining PGF; therefore, its incidence cannot be accurately determined. A consensus definition is required to help advance this field. Without a clear definition, it is impossible to critically analyze the safety and efficacy of interventions. Most interventions are symptomatic and fail to address the underlying etiologies, which are likely to be different in different persons. Given these complexities, more research is needed in this field to achieve advances in defining and resolving the problem of PGF.
Management of Non-infective Pulmonary Complications: What is New?
Non-infectious pulmonary complications (NIPCs) are an unsolved problem following allo-HCT. The etiology is multifactorial and includes pre-transplant chemotherapy and irradiation, infections, and GvHD16. Clinically, NIPCs are often experienced by patients with signs and symptoms of pneumonia as well as evidence of widespread alveolar damage in the absence of lower respiratory tract infections and cardiac, renal, or iatrogenic etiology17. Risk factors for developing NIPCs include previous infections, advanced age with impaired performance status prior to allo-HCT, conditioning intensity, total-body irradiation (TBI) in high doses, and high-grade GvHD18–20. The pathophysiology of NIPCs remains ill-defined and effective treatment approaches have not been standardized. The American Thoracic Society (ATS) recently proposed that NIPCs should be categorized anatomically into interstitial tissue, vascular endothelium, or airway epithelium disease16; however, diagnosing NIPCs according to these criteria remains difficult because affected patients are often too unwell to permit evaluation of the pulmonary lesion. In practice, NIPCs include idiopathic pneumonia syndrome (IPS) and late-onset non-infectious pulmonary complications (LONIPCs).
IPS is characterized by an acute lung dysfunction of non-infectious etiology with a median time of onset of 19 d (range 4-106 d) after allo-HCT19,20. In contrast, LONIPCs are major pulmonary events occurring > 3 months after allo-HCT in up to 20% of allotransplant recipients20, leading to various clinical sequelae and are often associated with increased morbidity and mortality.
IPS is usually associated with respiratory failure and mortality rates > 50%. TNF-α has been implicated in the pathophysiology of IPS. High-dose systemic steroids (2 to 4 mg/kg) and, in selected cases of refractory disease, the addition of etanercept, a soluble TNF-α-binding protein (0.4 mg/kg administered twice weekly up to a maximum of 8 doses) have been beneficial21–23.
After allo-HCT, two clinical situations lead to the diagnosis of LONIPCs: (1) development of respiratory symptoms, such as dyspnea, cough, sputum, and wheezing, or (2) deterioration of lung function based on sequential post-allo-HCT screening pulmonary function tests. Although lung biopsy is the gold standard for LONIPCs diagnosis, certain patients after allo-HCT can undergo lung surgery due to associated complications24,25. Thus, most LONIPC diagnoses rely mainly on PFT and CT scan data and are categorized as interstitial pneumonia, cryptogenic organizing pneumonia (COP), bronchiolitis obliterans syndrome (BOS), and lung fibrosis.
According to the 2005/2014 National Institutes of Health consensus criteria26, BOS is the only diagnostic manifestation of chronic pulmonary GvHD. A diagnosis of BOS requires at least one other distinctive manifestation of chronic GvHD in a separate organ system, excluding respiratory tract infections. Functional diagnosis criteria for BOS are as follows: ratio of forced expiratory volume in 1st second (FEV1)/forced vital capacity (FVC) < 0.7, FEV1 < 75% of the predicted value, and residual volume (RV) > 120% of the predicted volume27. Air trapping can be visualized on a high-resolution lung CT scan showing a mosaic pattern of intensified attenuation on expiratory cuts. Bronchial thickening, bronchiectasis, and centrilobular micronodules have also been associated with a diagnosis of BOS28.
To date, no treatment has been shown to cure BOS. Despite the absence of evidence for efficacy, systemic steroid administration is the standard therapy29. Currently, the development of more effective treatment strategies for BOS is needed, focusing on patients with early stage BOS who are likely to be more responsive to treatment. Several studies have assessed the efficacy of various immunosuppressive drugs including rituximab, bortezomib, extracorporeal photopheresis, iburutinib, and ruxolitinib with variable success30–32. In addition to these limited treatment options, pulmonary rehabilitation may be an important adjunctive therapy to improve patient's quality of life33. Lung transplantation is currently a suitable therapeutic option in selected allo-HCT recipients with post-BOS chronic respiratory failure34,35.
Overall, the pathophysiology of NIPCs remains ill-defined and effective treatment approaches have not been standardized. Moreover, the prognosis of NIPCs remains unclear. For example, the impact of the stem cell source (bone marrow versus peripheral blood versus umbilical cord blood) has not been fully evaluated as a cause for NIPCs, and the effect of the expanded use of peripheral blood and umbilical cord blood from unrelated donors on the incidence and severity of disease has not been elucidated. In addition, cooperation with long-term follow-up teams in screening and early detection strategies for LONIPCs should be revisited at each transplantation center.
TA-TMA-diagnosis, Risk Factors, and Treatment
Introduction
Complement is an elaborate system that participates in the innate immune response. Genetic variants and autoantibodies that lead to unregulated complement activation are implicated in the pathogenesis of various human diseases36. Paroxysmal nocturnal hemoglobinuria (PNH) is a prototypical model of complement activation and inhibition. Eculizumab, a first-in-class complement inhibitor, was initially approved for PNH, followed by approval for complement-mediated diseases/complementopathies across specialties, including atypical or complement-mediated hemolytic uremic syndrome (aHUS or CM-HUS), myasthenia gravis, and neuromyelitis optica spectrum disorder. In addition to revealing unmet needs and paving the way for next-generation complement therapeutics, preclinical and clinical studies have also delineated other diseases that might benefit from complement inhibition.
Transplant-associated Thrombotic Microangiopathy (TA-TMA)
TA-TMA is a life-threatening complication of allo-HCT and is observed in 7-39% of allo-HCT recipients. It manifests with the clinical triad of TMA: thrombocytopenia, microangiopathic hemolytic anemia, and organ damage (primarily involving the kidneys or central nervous system)38. Diagnosis is largely hindered by the high incidence of cytopenia and organ dysfunction in allo-HCT recipients. Currently used diagnostic criteria include the Bone Marrow Transplant Clinical Trials Network and the International Working Group criteria39,40. Both sets of criteria have been criticized for their diagnostic sensitivity. Therefore, the diagnosis remains complex due to the high incidence of cytopenia and organ dysfunction in allo-HCT, as well as the lack of sensitivity of the current diagnostic criteria32. In the pediatric population, the proposed severity criteria have incorporated a rough marker of complement activation (soluble C5b-9) to facilitate early diagnosis and treatment33.
Our understanding of TA-TMA pathophysiology is evolving rapidly. Initially, the syndrome was considered and treated as a form of thrombotic thrombocytopenic purpura (TTP). However, plasma exchange has a limited efficacy in these patients. In line with these findings, studies have shown that ADAMTS13 is not deficient in TA-TMA and, therefore, cannot be used as a disease marker. A better understanding of CM-HUS has helped researchers and clinicians understand that both pathophysiological and clinical aspects of TA-TMA resemble those of CM-HUS more than those of TTP. This notion is supported by the genetic and functional evidence of complement activation in TA-TMA. Jodele et al were the first to describe complement-related mutations in pediatric allo-HCT recipients34, with additional data on a poor prognosis in patients harboring APC-related mutations.
Genetic susceptibility data supports that the two-hit hypothesis is true for TA-TMA. The second hit may result from several clinical factors that have been associated with TA-TMA, including age, donor type, conditioning regimen, calcineurin or mTOR inhibitors, graft-versus host disease, or infections. Although several cross-sectional associations have been reported, only a few studies have investigated the underlying mechanisms. Notably, complement dysregulation has been implicated in GvHD regulation in mice and humans. Additional links between GvHD and complement activation have been demonstrated in human cutaneous tissues, where C3 inhibition by compstatin reduced CD4+ T-cell proliferation and Th1/Th17 polarization. A recent in vitro study also implicated the C5a/C5aR IL-17A axis in chronic GvHD. Furthermore, C3 levels have been associated with sclerotic cutaneous GvHD, and patients with sclerotic GvHD have shown abnormalities in complement factor H and APC functional assays. Complement activation has also been linked at thrombin generation in patients treated with antithymocyte globulin.
Indeed, accumulating genetic and functional data suggest increased complement activation and possible genetic predisposition in both the adult and pediatric population with TA-TMA35,36. In this complex setting of predisposing endothelial injury syndromes, other markers of endothelial damage, such as neutrophil extracellular traps, have also been found to increase and merit further study37,38. In this context, MASP-2 (mannan-binding lectin serine protease 2) has emerged as a potential therapeutic target based on its involvement in the complement pathway and direct interactions with the coagulation system. As in vitro data on MASP-2 levels in TA-TMA are very limited, experts do not support its use as a marker in this setting. Therefore, the potential role of the lectin pathway in TA-TMA requires further investigation.
In the pre-eculizumab era, the standard of care for TA-TMA involved cessation of calcineurin or mammalian target of rapamycin (mTOR) inhibitors, supportive treatment, corticosteroids, and sometimes plasma exchange or rituximab, according to each center's policy. Most patients with TA-TMA are refractory to conventional treatment, leading to high mortality rates, reaching 100% (median, 75%). In recent years, eculizumab has been increasingly used to treat both adult and pediatric patients with TA-TMA. Despite high response rates to eculizumab treatment (up to 93%), the overall survival remains low (approximately 30%) in early reports on the adult population. Notably, Jodele et al. achieved an increased 1-year survival rate of 66% in 64 pediatric eculizumab-treated patients compared to 17% in a historic control group. Although the results are encouraging compared to the mortality rates in the pre-eculizumab era, the timing of initiation, appropriate patient selection, dosing, and duration of therapy remain to be further investigated in this complex field of patient with transplants.
Regarding novel complement inhibitors, the C5 inhibitor conversin was successfully used in a patient with TA-TMA with a C5 variant that caused resistance to eculizumab treatment. Interestingly, a phase 2 single-arm, open-label study of the MASP-2 inhibitor narsoplimab in 19 patients with TA-TMA also reported an increased median overall survival compared to the historical control using conventional treatment (347 versus 21 days from TA-TMA diagnosis). This drug is currently under priority review by the FDA. It should be also noted that novel drugs approved for PNH are currently evaluated in TA-TMA: ravulizumab in a phase 3 trial of adult (NCT04543591) and pediatric (NCT04557735) patients with TA-TMA (NCT02946463), and pegcetacoplan in a phase 2 study (NCT05148299). Finally, coversin or nomacopan, a C5 inhibitor that also blocks leukotriene B4, was evaluated in a two-part phase 3 trial in pediatric TA-TMA (NCT04784455). A summary of these results is presented in Table 1.
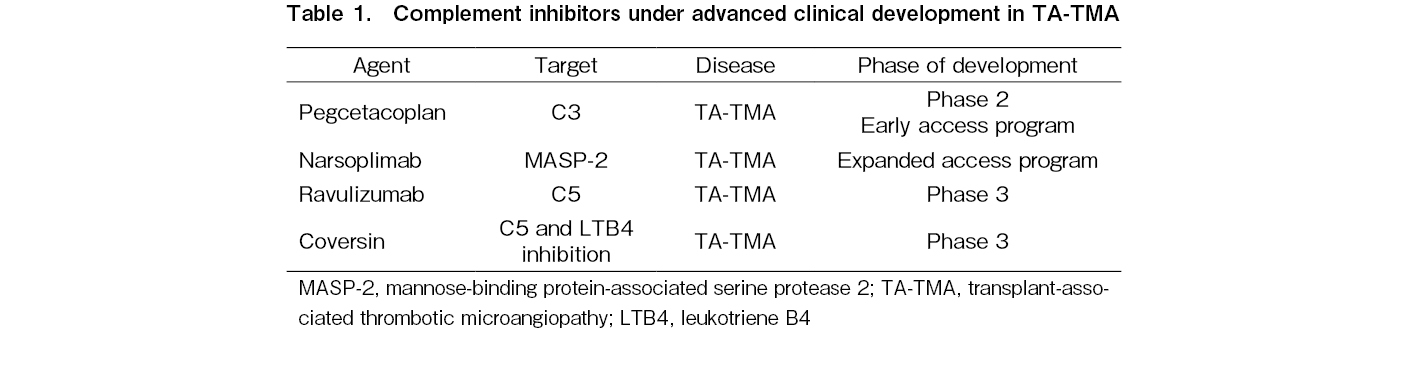
In conclusion, complement inhibition transformed TA-TMA from a lethal complication into a treatable syndrome. Further advances in the field are required to provide access to testing and enable early initiation of complement inhibition. Additionally, better understanding of the absence of response to eculizumab and personalized algorithms for novel inhibitors are required.
Conclusions
Post-allo-HCT complications, including graft dysfunction, pulmonary non-infectious complications, and TA-TMA, are common. They are difficult to diagnose and no standardized treatment approaches are available. Conditioning intensity modifications and the introduction of novel agents can ameliorate post-allo-HCT complications and improve post-transplant outcomes. The introduction of disease-specific novel agents can overcome some of the obstacles in the allo-HCT procedure. Future research is needed to improve our understanding and treatment of these complications.
Acknowledgments
By Dr. Eleni: Given the broad scope of this review, the authors often refer to specialized review articles rather than primary literature, and they have been able to include only selected examples of original work in the field. Therefore, the authors thank colleagues who are not specifically cited for their contribution and their understanding. E.G. is supported by the ASH Global Research Award 2020. Dr. Halahleh acknowledged prof. Robert Gale for editing the initial PGF manuscript.
Author Contributions
KH wrote PGF section and edited the whole manuscript; YA wrote post allo-HCT pulmonary complication section; and EG wrote the draft of TA-TMA. All authors read and approved the final manuscript.
Conflicts of Interest
EG has consulted for Amyndas, Alexion, and Omeros Pharmaceuticals, Inc. Other authors declare no conflicts of interest associated with this article. Disclosure forms provided by the authors are available on the website.
References
1.Valcárcel D, Sureda A, Graft Failure, Carreras E, Dufour C, Mohty M, and Kroger N, The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies, [Internet]. 7th edition, Springer, 2019; 307-13, https://www.ncbi.nlm.nih.gov/books/NBK553978/ [Accessed: 7 July 2022]
2.Lee KH, Lee JH, Choi SJ, Lee JH, Kim S, Seol M, et al. Failure of trilineage blood cell reconstitution after initial neutrophil engraftment in patients undergoing allogeneic hematopoietic cell transplantation – frequency and outcomes. Bone Marrow Transplant. 2004; 33: 729-34.
3.Shono Y, Shiratori S, Kosugi-Kanaya M, Ueha S, Sugita J, Shigematsu A, et al. Bone marrow graft-versus-host disease: evaluation of its clinical impact on disrupted hematopoiesis after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2014; 20: 495-500.
4.Dominietto A, Raiola AM, van Lint MT, Lamparelli T, Gualandi F, Berisso G, et al. Factors influencing haematological recovery after allogeneic haemopoietic stem cell transplants: graft versus-host disease, donor type, cytomegalovirus infections and cell dose. Br J Haematol. 2001; 112: 219-27.
5.Zhao Y, Gao F, Shi J, Luo Y, Tan Y, Lai X, et al. Incidence, risk factors, and outcomes of primary poor graft function after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2019; 25: 1898-907.
6.Prabahran A, Koldej R, Chee L, Ritchie D. Clinical features, pathophysiology, and therapy of poor graft function post-allogeneic stem cell transplantation. Blood Adv. 2022; 6: 1947-59.
7.Man Y, Lu Z, Yao X, Gong Y, Yang T, Wang Y. Recent advancements in poor graft function following hematopoietic stem cell transplantation. Front Immunol. 2022; 13: 911174.
8.Stasia A, Ghiso A, Galaverna F, Raiola AM, Gualandi F, Luchetti S, et al. CD34 selected cells for the treatment of poor graft function after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. 2014; 20: 1440-3.
9.Tefferi A, Vaidya R, Caramazza D, Finke C, Lasho T, Pardanani A. Circulating interleukin (IL)-8, IL-2R, IL-12, and IL-15 levels are independently prognostic in primary myelofibrosis: a comprehensive cytokine profiling study. J Clin Oncol. 2011; 29: 1356-63.
10.Lund TC, Liegel J, Bejanyan N, Orchard PJ, Cao Q, Tolar J, et al. Second allogeneic hematopoietic cell transplantation for graft failure: poor outcomes for neutropenic graft failure. Am J Hematol. 2015; 90: 892-6.
11.Halahleh K, Gale RP, Da'na W, Ma'koseh M, Saadeh S, Alan W, et al. Therapy of posttransplant poor graft function with eltrombopag. Bone Marrow Transplant. 2021; 56: 4-6.
12.Giammarco S, Sica S, Chiusolo P, Laurenti L, Sorá F, Martino M, et al. Eltrombopag for the treatment of poor graft function following allogeneic stem cell transplant: a retrospective multicenter study. Int J Hematol. 2021; 114: 228-34.
13.Nampoothiri RV, Ho L, McEwan C, Pasic I, Lam W, Law AD, et al. Efficacy and cost analysis of eltrombopag in thrombocytopenia and poor graft function post allogeneic hematopoietic cell transplantation. Bone Marrow Transplant. 2021; 56: 2471-6.
14.Shi MM, Kong Y, Song Y, Sun YQ, Wang Y, Zhang XH, et al. Atorvastatin enhances endothelial cell function in posttransplant poor graft function. Blood. 2016; 128: 2988-99.
15.Kong Y, Wang Y, Zhang YY, Shi MM, Mo XD, Sun YQ, et al. Prophylactic oral NAC reduced poor hematopoietic reconstitution by improving endothelial cells after haploidentical transplantation. Blood Adv. 2019; 3: 1303-17.
16.Soubani AO, Pandya CM. The spectrum of noninfectious pulmonary complications following hematopoietic stem cell transplantation. Hematol Oncol Stem Cell Ther. 2010; 3: 143-15.
17.Panoskaltsis-Mortari A, Griese M, Madtes DK, Belperio JA, Haddad IY, Folz RJ, et al. An official American Thoracic Society research statement: noninfectious lung injury after hematopoietic stem cell transplantation: idiopathic pneumonia syndrome. Am J Respir Crit Care Med. 2011; 183: 1262-79.
18.Cooke KR, Yanik G. Acute lung injury after allogeneic stem cell transplantation: is the lung a target of acute graft-versus-host disease?. Bone Marrow Transplant.
2004; 34: 753-65.
19.Fukuda T, Hackman RC, Guthrie KA, Sandmaier BM, Boeckh M, Maris MB, et al. Risks and outcomes of idiopathic pneumonia syndrome after nonmyeloablative and conventional conditioning regimens for allogeneic hematopoietic stem cell transplantation. Blood. 2003; 102: 2777-85.
20.Sakaida E, Nakaseko C, Harima A, Yokota A, Cho R, Saito Y, et al. Late-onset noninfectious pulmonary complications after allogeneic stem cell transplantation are significantly associated with chronic graft-versus-host disease and with the graft-versus-leukemia effect. Blood. 2003; 102: 4236-42.
21.Yanik G, Hellerstedt B, Custer J, Hutchinson R, Kwon D, Ferrara JL, et al. Etanercept (Enbrel) administration for idiopathic pneumonia syndrome after allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2002; 8: 395-400.
22.Chellapandian D, Lehrnbecher T, Phillips B, Fisher BT, Zaoutis TE, Steinbach WJ, et al. Bronchoalveolar lavage and lung biopsy in patients with cancer and hematopoietic stem-cell transplantation recipients: a systematic review and meta-analysis. J Clin Oncol. 2015; 33: 501-9.
23.Jagasia MH, Greinix HT, Arora M, Williams KM, Wolff D, Cowen EW, et al. National Institutes of Health Consensus Development Project on Criteria for Clinical Trials in Chronic Graft-versus-Host Disease: I. The 2014 Diagnosis and Staging Working Group report. Biol Blood Marrow Transplant. 2015; 21: 389-401.e1.
24.Cheng GS, Stednick ZJ, Madtes DK, Boeckh M, McDonald GB, Pergam SA. Decline in the use of surgical biopsy for diagnosis of pulmonary disease in hematopoietic cell transplantation recipients in an era of improved diagnostics and empirical therapy. Biol Blood Marrow Transplant. 2016; 22: 2243-9.
25.Filipovich AH, Weisdorf D, Pavletic S, Socie G, Wingard JR, Lee SJ, et al. National Institutes of Health consensus development project on criteria for clinical trials in chronic graft-versus-host disease: I. Diagnosis and staging working group report. Biol Blood Marrow Transplant. 2005; 11: 945-56.
26.Gunn ML, Godwin JD, Kanne JP, Flowers ME, Chien JW. High-resolution CT findings of bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. J Thorac Imaging. 2008; 23: 244-50.
27.Bergeron A. Late-onset noninfectious pulmonary complications after allogeneic hematopoietic stem cell transplantation. Clin Chest Med. 2017; 38: 249-62.
28.Tran J, Norder EE, Diaz PT, Phillips GS, Elder P, Devine SM, et al. Pulmonary rehabilitation for bronchiolitis obliterans syndrome after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2012; 18: 1250-4.
29.Gavriilaki E, de Latour RP, Risitano AM. Advancing therapeutic complement inhibition in hematologic diseases: PNH and beyond. Blood. 2022; 139: 3571-82.
30.Gavriilaki E, Brodsky RA. Complementopathies and precision medicine. J Clin Invest. 2020; 130: 2152-63.
31.Gavriilaki E, Anagnostopoulos A, Mastellos DC. Complement in thrombotic microangiopathies: unraveling ariadne's thread into the labyrinth of complement therapeutics. Front Immunol. 2019; 10: 337.
32.Gavriilaki E, Sakellari I, Anagnostopoulos A, Brodsky RA. Transplant-associated thrombotic microangiopathy: opening Pandora's box. Bone Marrow Transplant. 2017; 52: 1355-60.
33.Jodele S, Davies SM, Lane A, Khoury J, Dandoy C, Goebel J, et al. Diagnostic and risk criteria for HSCT-associated thrombotic microangiopathy: a study in children and young adults. Blood. 2014; 124: 645-53.
34.Jodele S, Licht C, Goebel J, Dixon BP, Zhang K, Sivakumaran TA, et al. Abnormalities in the alternative pathway of complement in children with hematopoietic stem cell transplant-associated thrombotic microangiopathy. Blood. 2013; 122: 2003-7.
35.Gavriilaki E, Chrysanthopoulou A, Sakellari I, Batsis I, Mallouri D, Touloumenidou T, et al. Linking complement activation, coagulation, and neutrophils in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2019; 119: 1433-40.
36.Gavriilaki E, Touloumenidou T, Sakellari I, Batsis I, Mallouri D, Psomopoulos F, et al. Pretransplant genetic susceptibility: clinical relevance in transplant-associated thrombotic microangiopathy. Thromb Haemost. 2020; 120: 638-46.
37.Jodele S, Zhang K, Zou F, Laskin B, Dandoy CE, Myers KC, et al. The genetic fingerprint of susceptibility for transplant-associated thrombotic microangiopathy. Blood. 2016; 127: 989-96.
38.Gloude NJ, Khandelwal P, Luebbering N, Lounder DT, Jodele S, Alder MN, et al. Circulating dsDNA, endothelial injury, and complement activation in thrombotic microangiopathy and GVHD. Blood. 2017; 130: 1259-66.
39.Ho VT, Cutler C, Carter S, Martin P, Adams R, Horowitz M, et al. Blood and marrow transplant-clinical trials network toxicity committee consensus summary: thrombotic microaniopathy after hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2005; 11: 571-5.
40.Ruutu T, Barosi G, Benjamin RJ, Clark RE, George JN, Gratwohl A, et al. Diagnostic criteria for hematopoietic stem transplant associated thrombotic microaniopathy: results of a consensus process by an international working group. Haematologica. 2007; 92: 95-100.
Search
News


