Volume 5 (2022) Issue 3 No.2 Pages 75-78
Abstract
Omenn syndrome (OS) is typically observed in the autosomal recessive form of severe combined immunodeficiency (SCID) with autoreactive manifestations, and it requires allogeneic hematopoietic stem cell transplantation. Unlike non-OS SCID, a conditioning regimen is usually required to eradicate T-cells; however, optimal conditioning regimens are not established mainly because of the rarity of OS. Here, we report a case of hematopoietic stem cell transplantation with a reduced dose of busulfan, as a conditioning regimen and successful engraftment with complete chimerism. OS was diagnosed in a one-month-old boy based on a diffuse erythematous rash, absent B-cells, and activated T-cells. Genetic analysis failed to identify causative mutations for OS/SCID, such as RAG1/2. Bone marrow transplantation was performed from his HLA-matched sister with a conditioning regimen consisting of targeted busulfan, fludarabine, and anti-thymocyte globulin. Cyclosporine had been administered before transplantation to control abnormal T-cell activation and continued for graft-versus-host disease (GVHD) prophylaxis. Engraftment was achieved on day 12, and no GVHD symptoms were observed. For stem cell transplantation for OS, prior control of autoreactive symptoms with immunosuppressants is important for safe transplantation and reduced intensity conditioning (RIC) can be an option to achieve sustained engraftment.
Introduction
Omenn syndrome (OS) is one of the form of severe combined immunodeficiency (SCID) with autoreactive manifestations. Due to immune dysregulation, patients with OS typically have erythroderma,
The patients with OS usually die from uncontrolled immune dysregulation due to recurrent infections before 6 months of age, unless they are treated with allogeneic hematopoietic stem cell transplantation (HSCT)1. The overall survival is significantly higher after HSCT from matched sibling donors (MSDs) than from any other donor type3. When an MSD is available, a conditioning regimen is not required to achieve engraftment in patients with non-OS SCID without T-cells. However, as OS has autoreactive leaky T-cells, a conditioning regimen should be used to eradicate the T-cells of a patient, which cause engraftment failure even in HSCT from MSD. With careful consideration of long-term complications and quality of life, reduced-intensity conditioning (RIC) regimens are recommended2,4–6. However, few reports have documented detailed transplantation for OS because of its rarity. Here, we report a case of HSCT with an RIC regimen and successful engraftment in an infant diagnosed with OS.
Case Presentation
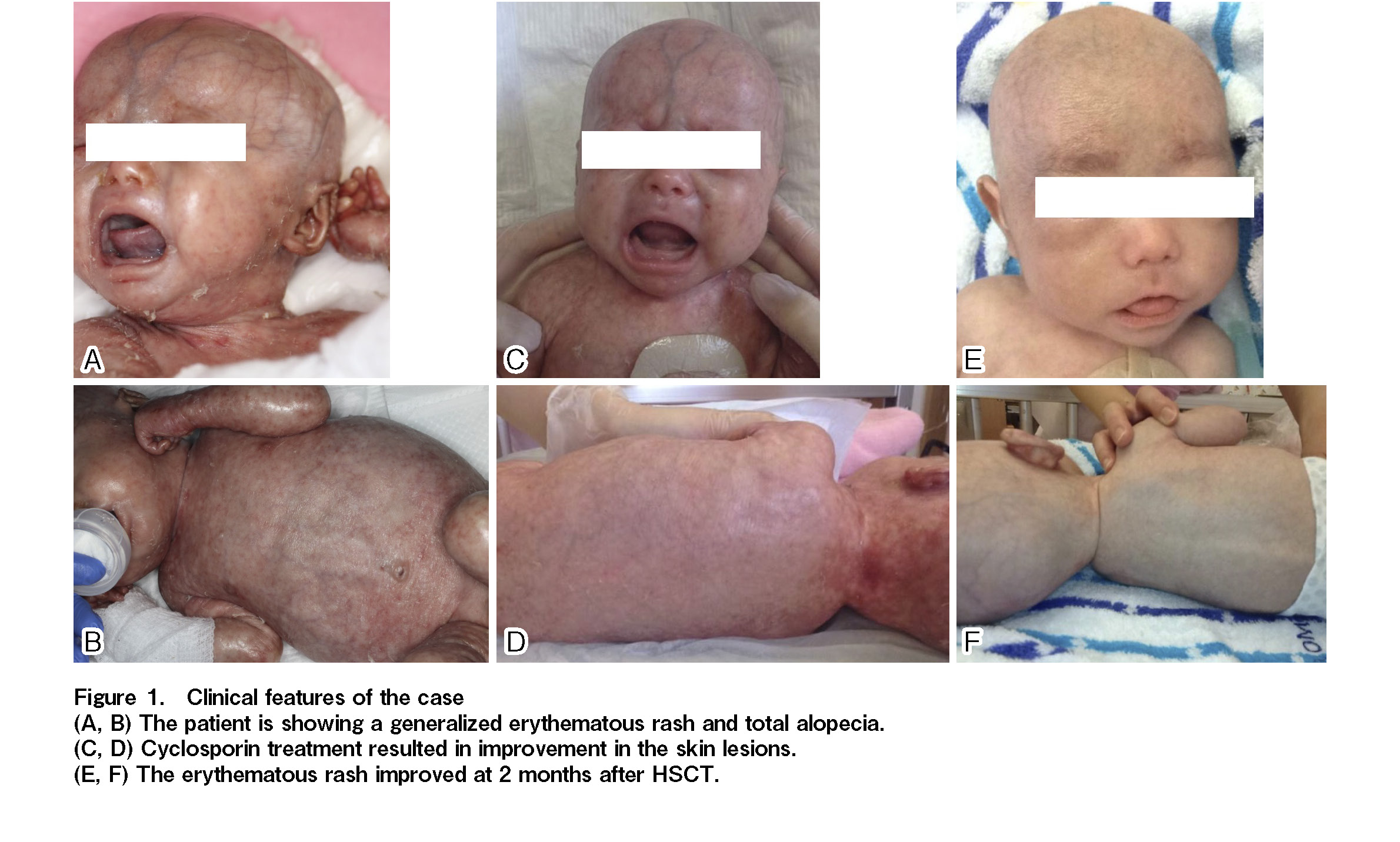
The patient, a one-month-old boy, was born at term and a diffuse erythematous rash was present from birth. Clinical history and examination showed exfoliative erythroderma, alopecia (Figure 1A and 1B), and fever on several occasions. Laboratory studies at day 0 demonstrated white blood cells (WBC) 19.1 × 103/μL, lymphocytes 0.67 × 103/μL, and neutrophils 15.6 × 103/μL. Flow-cytometric analysis revealed the absence of B-cells, and most lymphocytes were T-cells in the peripheral blood. Fluorescence in situ hybridization (FISH) of sex chromosomes showed that all T-cells originated from the patient, not the mother. Further detailed flow-cytometric analysis showed absence of thymic-naïve T-cells, while most of the T-cells (both CD4+ and CD8+) were HLA-DR positive. These data were consistent with the immune phenotype of OS.
The T-cell recombination excision circles (TREC) and kappa-deleting element recombinant cirle (KREC) copy numbers were 34 and 22 copies/μg DNA, respectively, which were extremely low, and his condition was diagnosed as OS; however, genetic analysis including RAG1/2 failed to identify the causative mutations for OS/SCID (
Based on the diagnosis of OS and immunological findings with autoreactive T-cells, cyclosporine had been administered orally before HSCT to control abnormal T-cell activation, which resulted in marked improvement in clinical symptoms, including an erythematous rash (Figure 1C and 1D). At two months, bone marrow transplantation was performed from an HLA-matched sister. The total number of infused cells was 13.8 × 108 cells/kg (CD34-positive cells 19.1 × 106/kg). The conditioning regimen consisting of fludarabine (30 mg/m2 per day) from day −7 to −2 and busulfan from day −3 to −2 (actual total AUC of 51.6 mg/L × h, four times a day) was combined with anti-thymocyte globulin (2.5 mg/kg per day) from day −7 to −6. Oral cyclosporine and a short course of methotrexate was administered for graft-versus-host disease (GVHD) prophylaxis. The clinical course is summarized in
Neutrophil engraftment was achieved on day 12. Engraftment of donor cells was confirmed using FISH analysis of sex chromosomes, reaching 97% as early as on day 14. After engraftment, the condition of the skin improved substantially (Figure 1E and 1F). No severe adverse events were observed during the transplantation. At day 52, chimerism analysis showed sustained full-donor engraftment for each lineage, including T-cells, B-cells, NK cells, monocytes, and neutrophils.
Currently, the patient is in good general condition with sustained complete donor chimerism without GVHD at one-year from transplantation, and cyclosporine was successfully discontinued. Flow-cytometric analysis showed an increased generation of thymic-naïve T-cells, and CD3+, CD19+, and CD56+ cells, one-year post-transplantation.
Discussion
We presented a case of an OS patient who was successfully treated with the RIC regimen. In HSCT for OS, abnormal autoreactive T-cells cause tissue damage in the skin, gut, and liver, which increases the risk of transplantation-related mortality and morbidity. Control of T-cell activation before HSCT reduces the risk of fatal complications7. Cyclosporine can suppress the nuclear factors of activated T-cell transcription and improve patient status as a bridge to HSCT8, as shown in our case.
Unlike typical SCID, a conditioning regimen is required to eliminate autoreactive cells in OS, even in HSCT from MSD. Considering the potential risks of acute and late complications9, RIC regimens should be adopted to minimize acute and late complications.
The standard dosage of busulfan is usually based on the pharmacokinetics of the test dose. Although the joint EBMT/ESID Inborn Errors Working Party recommends using a target area under the curve (60-70 mg/L×h)5, the optimal dosage of busulfan for patients with OS who receive RIC is uncertain, and there is room to optimize the use of busulfan at a target area under the curve.
More comprehensive methods, such as whole exome/genome sequencing, are required to identify a causative variant for SCID/OS patients without known causative variants.
The survival rate was the lowest for children with SCID who were older than 3.5 months of age and had active infection at the time of transplantation10. Early diagnosis and the advent of newborn screening for SCID will help improve survival.
We presented a case of a patient with OS who underwent reduced-intensity HSCT. The RIC regimen consisting of fludarabine and dose-adjusted busulfan was found to be an effective and safe treatment for OS. Further studies are required to further optimize HSCT for OS.
Acknowledgments
The authors would like to thank Drs. Kenichi Sakamoto, Shin-Ichi Tsujimoto, Meri Uchiyama, Yoshihiro Gocho, Takanori Mizuno, Masahiro Sugawa, Kazue Yoshida, and Yukio Matsumoto for patient management. The authors wish to thank the medical editor from the Division of Postgraduate Education and Training of the National Center for Child Health and Development for editing this manuscript. This study was supported in part by a grant from the National Center for Child Health and Development, grant numbers 2020A-1 and 2020B-2.
Author Contributions
Y.M. and T.U. collected and interpreted the data and wrote the manuscript; M.K. and M.O. designed the study, interpreted the data, and wrote the manuscript;
Acknowledgments
The authors would like to thank Drs. Kenichi Sakamoto, Shin-Ichi Tsujimoto, Meri Uchiyama, Yoshihiro Gocho, Takanori Mizuno, Masahiro Sugawa, Kazue Yoshida, and Yukio Matsumoto for patient management. The authors wish to thank the medical editor from the Division of Postgraduate Education and Training of the National Center for Child Health and Development for editing this manuscript. This study was supported in part by a grant from the National Center for Child Health and Development, grant numbers 2020A-1 and 2020B-2.
Informed Consent
Informed consent was obtained by all participants in this study.
Conflicts of Interest
The authors declare no conflict of interest. Disclosure forms provided by the authors are available on the website.
References
1.Aleman K, Noordzij JG, de Groot R, van Dongen JJ, Hartwig NG. Reviewing Omenn syndrome. Eur J Pediatr. 2001; 160: 718-25.
2.Wahadneh A, Bin-Dahman H, Habahbeh Z, Abu-Shukear M, Ajarmeh M, Zyood R, et al. Successful second bone marrow transplantation in Omenn's syndrome after bone marrow aplasia: A case report. Pediatr Transplant. 2012; 16: E43-8.
3.Haddad E, Logan BR, Griffith LM, Buckley RH, Parrott RE, Prockop SE, et al. SCID genotype and 6-month posttransplant CD4 count predict survival and immune recovery. Blood. 2018; 132: 1737-49.
4.Rossi G, Zecca M, Giorgiani G, Bonetti F, De Stefano P, Locatelli F. Non-myeloabiative stem cell transplantation for severe combined immunodeficiency – Omenn syndrome. Br J Haematol. 2004; 125: 406-7.
5.Lankester AC, Albert MH, Booth C, Gennery AR, Güngör T, Hönig M, et al. EBMT/ESID inborn errors working party guidelines for hematopoietic stem cell transplantation for inborn errors of immunity. Bone Marrow Transplant. 2021; 56: 2052-62.
6.Gozdzik J, Czogala W, Skoczen S, Krasowska-Kwiecien A, Wiecha O, Mordel A, et al. Rapid full engraftment and successful immune reconstitution after allogeneic hematopoietic stem cell transplantation with reduced intensity conditioning in Omenn syndrome. Pediatr Transplant. 2009; 13: 760-5.
7.Wootten V, Goulden N, Veys P, Qasim W. A double cure: Omenn syndrome and β thalassaemia successfully treated with mismatched unrelated donor transplantation. Bone Marrow Transplant. 2012; 47: 589-90.
8.Meyer-Bahlburg A, Haas JP, Haase R, Eschrich U, Wawer A, Frank L, et al. Treatment with cyclosporin A in a patient with Omenn's syndrome. Arch Dis Child. 2002; 87: 231-3.
9.Caglayan Sozmen S, Isik S, Arikan Ayyildiz Z, Yildiz K, Cakır Y, Ozer E, et al. Cyclosporin treatment improves skin findings in Omenn syndrome. Pediatr Dermatol. 2015; 32:
10.Heimall J, Puck J, Buckley R, Fleisher TA, Gennery AR, Neven B, et al. Current Knowledge and Priorities for Future Research in Late Effects after Hematopoietic Stem Cell Transplantation (HCT) for Severe Combined Immunodeficiency Patients: A Consensus Statement from the Second Pediatric Blood and Marrow Transplant Consortium International Conference on Late Effects after Pediatric HCT. Biol Blood Marrow Transplant. 2017; 23: 379-87.
11.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000-2009. N Engl J Med. 2014; 371: 434-46.
Search
News


