Volume 5 (2022) Issue 3 No.1 Pages 69-74
Abstract
Allogeneic hematopoietic stem cell transplantation (HSCT) is a feasible treatment option for Gaucher disease (GD). Among 60 patients diagnosed with GD over 15 years (2004-2019), three children who underwent HSCT (January-November 2017) were analyzed. Two boys (cases 1 and 2) and one girl (case 3) received HSCT at 3, 7, and 10 years of age, respectively. Cases 1 and 3 received haplo-HSCT, while case 2 received HLA-identical related-donor transplantation. The CD 34 cell dose was 5-10×106/kg. Neutrophil and platelet engraftment were between days +14 to +21 and days +15 to +76. Post-HSCT chimerism was a 100% donor. None of the patients developed acute or significant chronic graft versus host disease (GVHD). All patients had febrile episodes with negative blood cultures. Major post-HSCT complications included EBV-viremia and recurrent lobar pneumonia in case 1, delayed engraftment and pure red cell aplasia (PRCA) in case 2, and pericardial effusion with tamponade in case 3. At a median of 49 months post-HSCT, all patients were stable with improved growth, absent organomegaly, and had completed immunization. The median cost of treatment was $23,038.96, which is 10.7%-13% of the yearly enzyme replacement therapy (ERT) cost. In a resource-limited setting like India, ERT is a financial burden and not a sustainable option. With improved treatment outcomes, haplo-HSCT is now a possible option for almost every patient, even if no HLA-identical donor is identified.
Introduction
Gaucher disease (GD), the most common lysosomal storage disorder (LSD), is inherited as an autosomal recessive trait and is characterized by the accumulation of glucocerebroside in the lysosomes, commonly in tissue macrophages, due to a deficiency of the enzyme glucocerebrosidase (lysosomal acid M-glucosidase)1. The glucocerebrosidase gene (GBA) has been mapped to chromosome 1, band q21, and more than 200 disease-causing mutations have been documented1. The incidence of GD in the general population is less than 1 in 30,000-40,000, with a higher prevalence (1 in 1,000) in patients from Ashkenazi Jewish descent2,3.
Three types of GD have been identified. Type 1 GD, a non-neuronopathic form, and the most common form (90%-95% of cases) with onset in adulthood. Types 2 and 3 are very rare and are neuronopathic forms with onset during infancy (type 2) or in the first decade of life (type 3), occurring in less than 1 in 100,000 of the population1–3. The hallmark of GD is glucocerebroside-laden macrophages (Gaucher cells) that accumulate throughout the body, leading to multi-systemic disease manifestations including thrombocytopenia, anemia, hepatosplenomegaly (HSM), bone disease, growth retardation, and neurological disorders4,5.
Disease-specific management of GD includes enzyme replacement therapy (ERT) and substrate reduction therapy (SRT) with excellent safety and efficacy6. ERT has proven to be the standard of care in the therapeutic management of symptomatic patients with GD, except in those with type 2 or severe type 3 GD, as the enzyme is unable to cross the blood-brain barrier (BBB). SRT with Miglustat (Zavesca, N-butyl deoxynojirimycin, OGT 918) licensed for patients who are unable to receive ERT inhibits glucosylceramide synthase, preventing the synthesis of glucosylceramide. This drug is able to cross the BBB, and its efficacy in the treatment of types 2 and 3 GD is being investigated3.
Experience with hematopoietic stem cell transplantation (HSCT) in GD is limited and the procedure is currently not in general use6. A recent review7 on the role of HSCT for GD has suggested that transplantation would represent a very safe curative approach, offering one-time complete correction of the disease, in contrast to the lifelong need for ERT with its associated expense and dependence on sophisticated drug manufacturing. The choice between HSCT and conservative measures depends on the assessment of the long-term benefits of HSCT over the significant mortality and morbidity associated with the procedure as well as resource availability.
Although a fair number of children with GD are referred to our institution, ERT or SRT is not an option due to resource constraints. The only option available to them is symptomatic therapy with splenectomy or the one-time curative option of HSCT. As most families do not have health insurance coverage, the onus lies on the family to arrange finances for medical care. Most of the families belong to the middle-income category, and the finances are arranged mainly through government (Prime Minister relief fund) and non-government organizations (Ratan Tata relief fund, and other private agencies). In this paper, we report the experience of allogeneic HSCT in our center and its cost-effectiveness when compared to ERT.
Patients and Methods
Among the 60 patients diagnosed with GD over 15 years (2004-2019) in our center, the three children who underwent HSCT for type 1 GD were retrospectively analyzed. The study protocol was approved by the Institutional Review Board (IRB) of Christian Medical college, Vellore, India, and the protocols used in the study were approved by Office of research, Institutional Review Board, Christian Medica college, IRB Min. No. 13500/Retro/dated: 28.10.2020. All three patients underwent HSCT between January 2017 and November 2017 after obtaining informed consent.
The source of the graft was granulocyte colony-stimulating factor (GCSF) mobilized peripheral blood stem cells in all patients. Conditioning consisted of a treosulfan (Treo; 42 G/m2 over 3 days), thiotepa (Thio; 8 mg/kg for one day), and fludarabine (Flu; 160 mg/m2 over 4 days)-based regimen with additional thymoglobulin (4.5 mg/kg over 3 days) for matched related HSCT, and single fraction (200cGy) TBI on day −1 for haploidentical HSCT (haplo-HSCT). Graft versus host disease (GVHD) prophylaxis included intravenous cyclosporine (CSA) (5 mg/kg/day in 2 divided doses from day −3 with regular monitoring of drug levels) and reduced-dose short course methotrexate (10 mg/m2 on day +1 and 7 mg/m2 on days +3, +6, and +11 post-HSCT) when the donor was HLA identical and post-transplant cyclophosphamide (100 mg/kg over 2 days on day +3 and +4), mycophenolate mofetil (MMF; 15 mg/kg thrice daily), and tacrolimus (0.02 mg/kg twice daily; dose adjusted with regular drug levels monitoring to target 5-10 ng/mL) from day +5 post-HSCT onwards when the donor was haploidentical. GVHD was prospectively recorded and graded using the Glucksberg classification (
Myeloid engraftment (neutrophil recovery) was defined as an absolute neutrophil count (ANC) of ≥500×109/L for 3 consecutive days after nadir and platelet engraftment as a platelet count of >20,000×109/L, independent of platelet transfusions for at least 7 consecutive days. Chimerism analysis was performed on genomic DNA extracted from whole blood on day +28 post-HSCT and then on days +60 and +100, and subsequently only if indicated as evidenced by cytopenia. Qualitative analysis of chimerism was performed using agarose or polyacrylamide gel electrophoresis (PAGE). Quantitative analysis was performed using capillary gel electrophoresis using an ABI 3130 or 3500 genetic analyzer with GeneScan 3.1 software (Applied Biosystems, Foster City, CA, USA)8,9.
GVHD prophylaxis was continued (MMF was stopped on day +50, while CSA or tacrolimus was continued until 6 months post-HSCT and then subsequently tapered and stopped over the next 6 months if there was no GVHD), as was antimicrobial prophylaxis (antiviral therapy with acyclovir for 3-4 months and co-trimoxazole and penicillin until 1 year post-HSCT). Immunization was initiated at 1-year post-HSCT, 1 month after stopping immunosuppressive therapy.
Results and Discussion
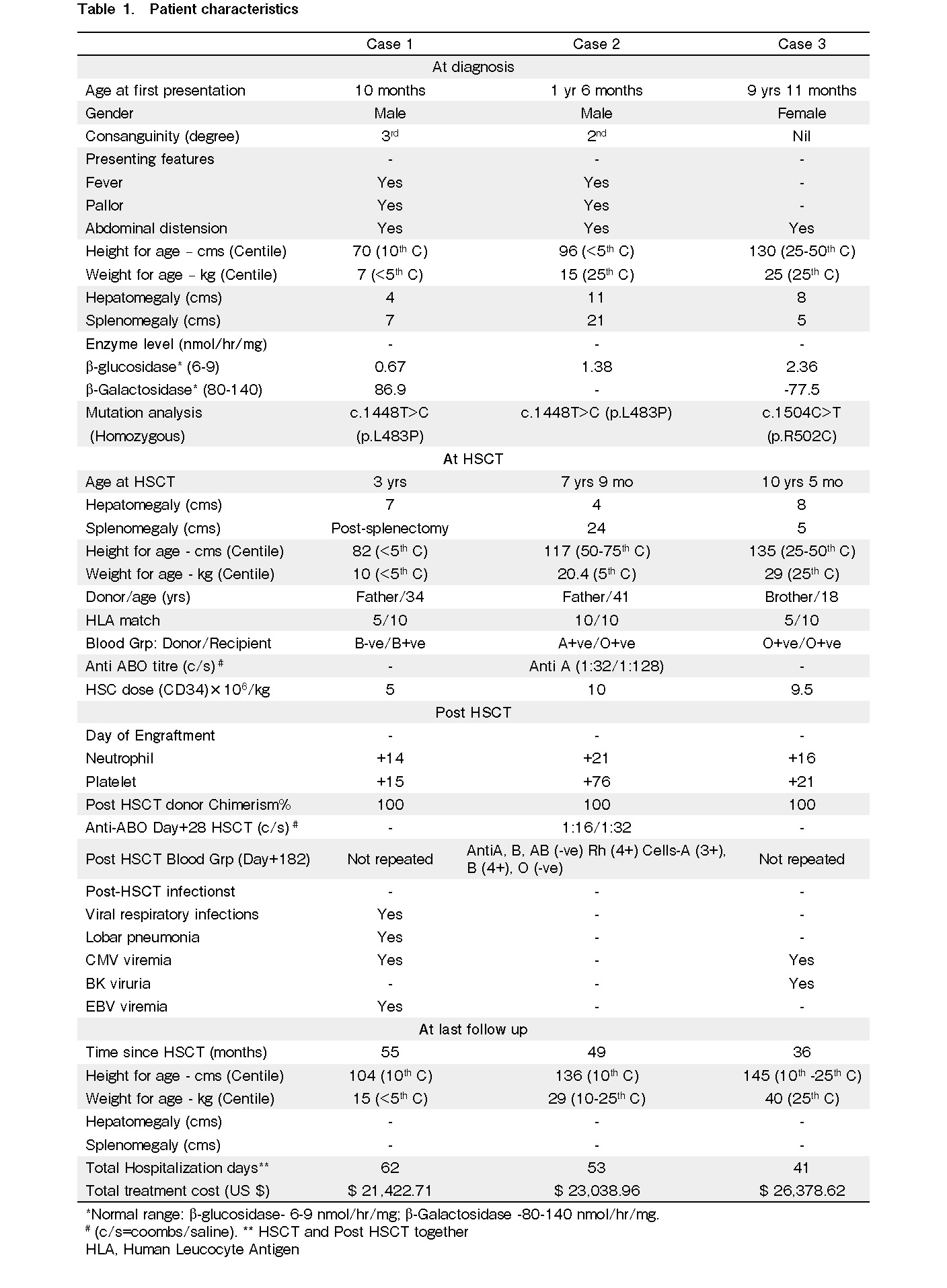
All three patients exhibited successful engraftment with 100% donor chimerism post-HSCT. The delayed engraftment in case 2 was likely due to massive splenomegaly and hypersplenism. The common peri-transplant complications were febrile neutropenia and mucositis. Case 2, who had a major blood group mismatched HSCT (Group A to O), developed pure red cell aplasia (PRCA) by day +25 post HSCT with positive serology (IgM) for parvovirus. He was treated with intravenous immunoglobulin (IVIg) and subsequently responded to steroids by day +324. Case 3 developed pericardial effusion with tamponade, which was likely a regimen-related (cyclophosphamide) toxicity that resolved with supportive measures and pericardiocentesis. In the post-HSCT period, case 1, who underwent HSCT post-splenectomy, required multiple hospitalizations for infective episodes (viral and bacterial). None of the patients were diagnosed with acute or significant chronic GVHD. Immunosuppressants were tapered and stopped by 1-year post-HSCT in all patients except case 2, where steroids were continued for a longer period for PRCA. At follow-up of 55, 49, and 36 months post-HSCT, all three children were stable with remarkably improved growth, off IST, with no GVHD, and had completed immunization. Case 1, with post-splenectomy status, was continued on penicillin prophylaxis. The organomegaly gradually resolved and was absent during the last follow-up. A repeat enzyme assay was not performed in any of these patients in the post-HSCT period. The median overall cost of treatment (including HSCT and post-HSCT) in these three cases was 23,038.96 US$ (21,422.71-26,378.62 US$), which amounted to only 10.7%-13% of the annual cost for ERT.
In patients with GD, at present, ERT is the recommended intervention of choice and is most often used. As patients are born with a deficient enzyme, this treatment modality by default must be lifelong. Considering the exuberant cost of ERT (amounting to $200,000 or more yearly), this is financially demanding on the family as well as the country, especially in developing countries such as India, with a population of approximately 1.5 billion and a GDP per capita income of $2,104 per year. Allogeneic HSCT is the only curative treatment that can provide a permanent source of the enzyme to patients with GD and is considerably less expensive than ERT. However, mortality and morbidity associated with HSCT, as well as the non-availability of HLA-matched donors, are major limitations.
We analyzed the outcomes of three children who underwent allogeneic HSCT in GD at our institution. The baseline patient characteristics, HSCT details, and post-HSCT features are described in Table 1 and the growth patterns in Figure 1. Only one patient had an HLA-identical related donor available; the other two underwent haploidentical donor HSCT. Early presentation in infancy was noted in case 1, while case 2 exhibited early childhood, and case 3, late childhood presentation. Case 1 underwent splenectomy due to massive splenomegaly and hypersplenism before HSCT. The extended period between diagnosis and HSCT was mainly due to the delay in arranging finances for HSCT. Most families belong to the middle-income category, and finances are arranged mainly through government and non-government organizations.
Recent advances in the field of transplantation have expanded the source of stem cells to haploidentical donors and have also reduced the risks associated with the procedure. These refinements have improved the outcome of haplo-HSCT, thus making allogeneic HSCT a possibility for every patient with GD, with a potential stem cell donor available for all children in their parents. Therefore, it is important to re-evaluate the risks and benefits of HSCT in comparison with the more conservative but noncurative options such as ERT and SRT in the treatment of GD; however, the lack of long-term outcome reports on HSCT in GD is a major limitation. A Cochrane systematic review did not find trials that were eligible for inclusion, although 32 were excluded3. A few case series have shown favorable clinical outcomes with a growth spurt, reversal of organomegaly, and a possible regression of skeletal changes and no further neurological deterioration in type 3 GD, with successful engraftment post-HSCT3. A non-randomized study by Young et al. suggested that HSCT (n=8) may be more effective than ERT (n=8) in reducing total body stores of glucocerebrosides10. Reports on unrelated umbilical cord blood stem cell transplantation (UCBT) in three children with GD from China11 showed success in two of them. In an article by Ito and Barettt7 on the review of 50 patients with GD who were treated with allogenic HSCT, the disease-free survival rate was reported to be 85%. However, in our series of patients, none of the patients had post-HSCT glucocerebroside levels evaluated.
We conclude that in a resource-limited setting such as India, ERT is a financial burden and not a sustainable long-term option, although it is the standard of care. Interestingly, in our series, in two out of the three children, stem cell donors were haploidentical related donors, and all the three children who underwent HSCT at our institute are living healthily. Due to its one-time cost and promising results, HSCT is worth considering as a definite treatment option for GD in countries such as India. With improved outcomes of haplo-HSCT, allogeneic HSCT is now a possible option for almost every patient, even if no HLA identical donor is identified. The major obstacle in implementing this curative one-time option for all patients with GD in a country such as India is the financial barricade. The major limitation of this study is the lack of long-term outcome data comparing the efficacy of HSCT in relation to ERT/SRT.
Acknowledgments
All patients and their families and all staff and colleagues of the department of Haematology.
Author Contributions
FNA and AA designed the study, clinical data accrual, and wrote paper. FNA, UPK, AK, AJD, SS, SL, AS, BG, and AA performed the research and analyzed data. ES performed molecular tests.
Conflicts of Interest
The authors declare no conflicts of interest. Disclosure forms provided by the authors are available on the website. AS is a member of the Editor of Blood Cell Therapy. He is not involved in the editorial evaluation or the decision to accept this article for publication.
References
1.Jmoudiak M, Futerman AH. Gaucher disease: pathological mechanisms and modern management. Br J Haematol. 2005; 129: 178-88.
2.Mehta A. Epidemiology and natural history of Gaucher's disease. Eur J Intern Med. 2006; 17: S2-5.
3.Somaraju UR, Tadepalli K. Hematopoietic stem cell transplantation for Gaucher disease. Cochrane Cystic Fibrosis and Genetic Disorders Group, editor. Cochrane Database Syst Rev [Internet]. 2017 Oct 18;
4.Beutler E. Gaucher disease: multiple lessons from a single gene disorder. Acta Paediatr. 2006; 95: 103-9.
5.Cox TM, Schofield JP. 3 Gaucher's disease: clinical features and natural history. Baillières Clin Haematol. 1997; 10: 657-89.
6.Puri RD, Kapoor S, Kishnani PS, Dalal A, Gupta N, Muranjan M, et al. Diagnosis and Management of Gaucher Disease in India – Consensus Guidelines of the Gaucher Disease Task Force of the Society for Indian Academy of Medical Genetics and the Indian Academy of Pediatrics. Indian Pediatr. 2018; 55: 143-53.
7.Ito S, Barrett AJ. Gauchers Disease―A Reappraisal of Hematopoietic Stem Cell Transplantation. Pediatr Hematol Oncol. 2013; 30: 61-70.
8.Alizadeh M, Bernard M, Danic B, Dauriac C, Birebent B, Lapart C, et al. Quantitative assessment of hematopoietic chimerism after bone marrow transplantation by real-time quantitative polymerase chain reaction. Blood. 2002; 99: 4618-25.
9.Sellathamby S, Balasubramanian P, Sivalingam S, Shaji RV, Mathews V, George B, et al. Developing an algorithm of informative markers for evaluation of chimerism after allogeneic bone marrow transplantation. Bone Marrow Transplant. 2006; 37: 751-5.
10.Young E, Chatterton C, Vellodi A, Winchester B. Plasma chitotriosidase activity in Gaucher disease patients who have been treated either by bone marrow transplantation or by enzyme replacement therapy with alglucerase. J Inherit Metab Dis. 1997; 20: 595-602.
11.Tang X, Luan Z, Wu N, Zhang B, Jing Y, Du H, et al. Treatment of Gaucher disease with allogeneic hematopoietic stem cell transplantation: report of three cases and review of literatures. Zhonghua Er Ke Za Zhi. 2015; 53: 810-6.
Search
News


