Volume 5 (2022) Issue 2 No.2 Pages 45-53
Abstract
Background: Hematopoietic stem cell transplantation (HSCT) provides curative therapy in almost 90% of patients with severe aplastic anemia (SAA). Older age, long duration of disease with consequent heavy exposure to transfusion, and active infection at the time of HSCT have a negative influence on the outcomes, causing graft failure (GF) and graft versus host disease (GVHD).
Purpose: To describe the outcomes of all patients with SAA who received hematopoietic stem cell transplantation at a tertiary center in Malaysia.
Materials and methods: We included a 20 y cohort of patients who underwent transplantation from January 1, 1999 to December 31, 2019. Data were obtained from electronic medical records. Demographics, clinical characteristics, and treatment outcomes were analyzed using descriptive statistics. Overall survival (OS) was analyzed using Kaplan-Meier curves. All analyses were conducted using the Statistical Package for the Social Sciences (SPSS) version 25.
Results: Eighty patients were analyzed. The median age at diagnosis was 19 years, and 59% patients were male (n = 47). Malay ethnicity was the highest (52.5%), followed by Chinese (20.0%) and Native Sabah (15.0%). The median duration from diagnosis to transplantation was 13.5 weeks. A majority of patients received Cy-ATG conditioning (n = 51, 63.8%). Forty-one patients (51.2%) used peripheral blood as stem cell source, 36 patients (45.0%) used granulocyte colony stimulating factor (G-CSF) primed marrow graft and 3 patients (3.8%) used both. The mean nucleated mononuclear cell and CD34 cell doses were 4.7 ± 1.7 × 108/kg and 4.6 ± 1.9 × 106/kg, respectively. Median engraftment for WBCs and platelets was 14 and 15 days, respectively. There was no difference in WBC and platelet engraftment in patients who received peripheral blood stem cell transplantation or bone marrow transplant. At a median follow-up of 54 months, 49 patients (61.3%) achieved complete remission and 8 patients (10.0%) achieved partial remission. The estimated 5 y OS was 63% and higher among those who received HSCT within 3 months of diagnosis. Twenty-two patients (27.5%) died within 100 d of transplantation, and a majority of these died due to pre-engraftment death.
Discussion and conclusions: Our study found that patients who received early allogeneic transplantation for SAA had better outcomes. Pre-engraftment failure was the major cause of transplant-related mortality within 100 d. Further studies are required to identify the factors responsible for delaying transplantation to improve treatment outcomes.
Introduction
Aplastic anemia (AA) is a bone marrow (BM) failure disorder characterized by pancytopenia and hypocellular marrow, with the absence of abnormal infiltrate or increased reticulin in the BM. It is a rare hematological disorder associated with significant morbidity and mortality1,2. The incidence of AA in western countries is approximately two per million per year and is estimated to be two- to three-fold higher in Asia. According to a retrospective analysis conducted between January 1993 and March 1996, AA is relatively common in Malaysia. Peak incidence is usually observed in the elderly group (8.6 per million), followed by a second peak in the young adolescent group (7.9 per million)3–6. Environmental factors, such as drugs, toxins, and chemicals may influence the incidence of AA.
AA can be categorized as congenital or acquired. The inherited form is rare, including Fanconi anemia, dyskeratosis congenita, diamond blackfan anemia, and shwachman-diamond syndrome. Immune destruction of hematopoietic stem cells plays an important role in the pathogenesis of acquired AA7–11. Therapeutic decisions are based on the severity of AA. At present, clinical practice guidelines are available to aid in therapeutic decision making12. Before immunosuppressants were widely used, severe AA (SAA) was associated with a high mortality rate, with most patients dying from hemorrhage and infection.
Recent treatment options, including allogeneic hematopoietic stem cell transplantation (HSCT) and immunosuppressive therapy (IST), mainly anti-thymocyte globulin (ATG) with cyclosporine (CYA), have resulted in better outcomes in AA patients. Allogeneic HSCT from a Human Leukocyte Antigen (HLA)-matched sibling donor (MSD) is the preferred first-line treatment option for younger patients (< 40 years of age) with SAA or very severe AA (VSAA), as it offers restoration of normal hematopoietic cells from the donor and potential cure from the disease. Allogeneic transplantation can also be considered in SAA patients aged 41-60 years, who are deemed fit and have failed first-line immunosuppressive therapy. IST is the only option for patients who are ineligible for transplantation or those without a suitable donor13,14.
Until recently, there was a lack of data pertaining to the disease characteristics and outcomes of patients with SAA who underwent allogeneic HSCT in Malaysia. This study was undertaken to understand the demographic characteristics and outcomes of patients with SAA who underwent allogeneic transplantation. Understanding the factors affecting treatment outcomes is important for improving overall survival (OS) and long-term management in this group of patients.
Materials and Methods
Study design and population
This was a retrospective study involving all patients with SAA who underwent allogeneic HSCT at our center, Hospital Ampang between January 1999 and December 2019. Our center is the main public hospital for stem cell transplants in Malaysia. Most patients in Malaysia are referred to this center for transplantation. Sociodemographic data, clinical characteristics, complications, treatment regimens, and outcomes were collected from electronic medical records using a standardized data collection form. We included all SAA patients who underwent allogeneic HSCT at our center and who were diagnosed as having SAA based on the modified Camitta criteria15: BM biopsy cellularity < 25% (or 25%-50% with < 30% residual hematopoietic cells) together with two of the following three criteria: hemoglobin < 10 g/dL, reticulocytes < 50 × 109/L or < 1%; platelets < 50 × 109/L; and absolute neutrophil count (ANC) < 1.0 × 109/L. Patients with alternative causes of BM aplasia, hypoplastic MDS, and inherited BM failure syndrome were excluded from this study. The study was conducted in accordance with the Declaration of Helsinki and was approved by the Medical Research and Ethics Committee (MREC), Ministry of Health, Malaysia.
Sociodemographic characteristics, clinical and treatment outcomes
Sociodemographic data, clinical characteristics, complications, treatment regimens, and outcomes were reviewed from the medical records and transplantation database. Patient ethnicity was defined according to the national identification documents (Malay, Chinese, Indian, Native Sabah, Native Sarawak, and Native Semenanjung). Other additional information analyzed included age and sex of the patient and donor, Cytomegaloviurs (CMV) status, ABO grouping for patient and donor, interval between diagnosis and transplantation, engraftment parameters, transplant complications, and treatment outcomes. The infused cell dose, including nucleated mononuclear cells (NMC) for marrow grafts and CD34 cells for peripheral blood stem cell transplantation (PBSC) source were recorded where available. Patients received either fludarabine, cyclophosphamide, and ATG (Flu-Cy-ATG), or cyclophosphamide and ATG (Cy-ATG) as their conditioning regimen depending on the stem cell source, number of red cells transfused, and duration of illness prior to transplantation. The Flu-Cy-ATG conditioning regimen consisted of fludarabine (30 mg/m2) administered intravenously (IV) once daily from days −8 to −4, cyclophosphamide (60 mg/kg) administered IV once daily from days −3 to −2, and horse ATG (30 mg/kg) administered IV once daily from days −3 to −1. The Cy-ATG regimen consisted of cyclophosphamide (50 mg/kg) administered IV once daily from days −5 to −2 and horse ATG (30 mg/kg) administered IV once daily from days −4 to −2. All patients received standard graft versus host disease (GVHD) prophylaxis with cyclosporine (CYA) (1.5 mg/kg) twice a day from day −1 onward and methotrexate (8 mg/m2) on days +1, +3, +6, and +11. Remission status for all patients was assessed at the time of last review or death and defined according to the established guidelines14, 15. Diagnosis and grading of acute GVHD (aGVHD) and chronic GVHD (cGVHD) were documented according to the established criteria16. The time in days to the first of three consecutive days of white blood cell count (WBC) > 0.5 × 109/L and platelet count > 20 × 109/L were set as the engraftment parameters. Non-engraftment was defined when these two conditions were not observed after day +99. OS was calculated from the time of transplantation to death or the date of the last follow-up, as appropriate.
Statistical analysis
Data analysis was conducted using the Statistical Package for the Social Sciences (SPSS) version 25.0. Descriptive statistics were presented as frequencies, percentages, ranges, means, and medians. Associations between categorical variables were analyzed using the chi-square test. Independent t-tests were used to analyze the correlation between the two continuous variables. The Kaplan-Meier method was used to estimate median survival times, and the log-rank test at a 5% significance level was used to test the equality of survival between groups. Statistical significance was set at p < 0.05.
Results
Patient characteristics
Between January 1, 2009 and December 31, 2019, we identified 80 confirmed SAA patients who underwent allogeneic HSCT. A total of 47 patients (59%) were male, and the median age at diagnosis for each sex was 19 years (range: 15-65 years). Of these, Malay ethnicity was the highest (n = 42, 52.5%), followed by Chinese (n = 20, 25.0%) and Native Sabah (n = 12, 15.0%). Most of the transplantations were referred from Selangor (n = 24, 30%) and Sabah (n = 19, 23.8%). All patients showed a good performance status prior to transplantation, with a Karnofsky score ≥ 80. The interval from diagnosis to transplantation ranged from 2 weeks to 11.9 months (median 13.5 weeks).
Most patients received some form of treatment (i.e., ATG, CYA, tacrolimus, and/or eltrombopag) prior to transplantation and were transfusion-dependent (n = 52, 65%). Seventy-nine patients received HLA-matched sibling transplants, and one patient received a matched unrelated donor (MUD) transplant. A majority of the patients were conditioned with Cy-ATG (n = 51, 63.8%) and the remaining were administered Flu-Cy-ATG. Forty-one patients (51.2%) used peripheral blood as a stem cell source, 36 patients (45%) used granulocyte colony stimulating factor (G-CSF)-primed marrow grafts, and 3 patients (3.8%) used both. The mean nucleated mononuclear cell (NMC) and CD34 cell doses were 4.7 ± 1.7 × 108/kg and 4.6 ± 1.9 × 106/kg, respectively. The demographic and clinical characteristics of the patients are summarized in Table 1.

Haematopoietic recovery and treatment response
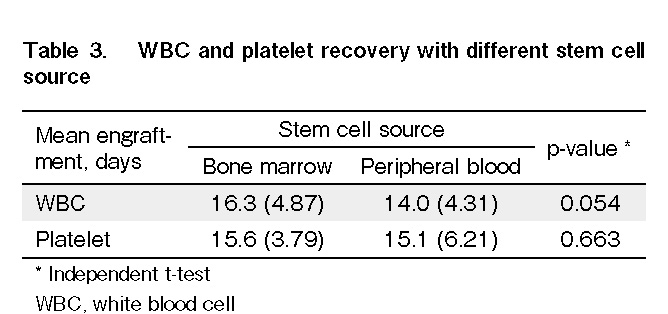
Engraftment parameters were evaluated in 64 patients. Fifteen patients died before engraftment. The median time to WBC and platelet engraftment in all patients was 14 d (range: 6-29 d) and 15 d (range: 8-35 d), respectively (Table 2). There was no significant reduction in days to WBC and platelet recovery in PBSC as compared to those in BM (Table 3).
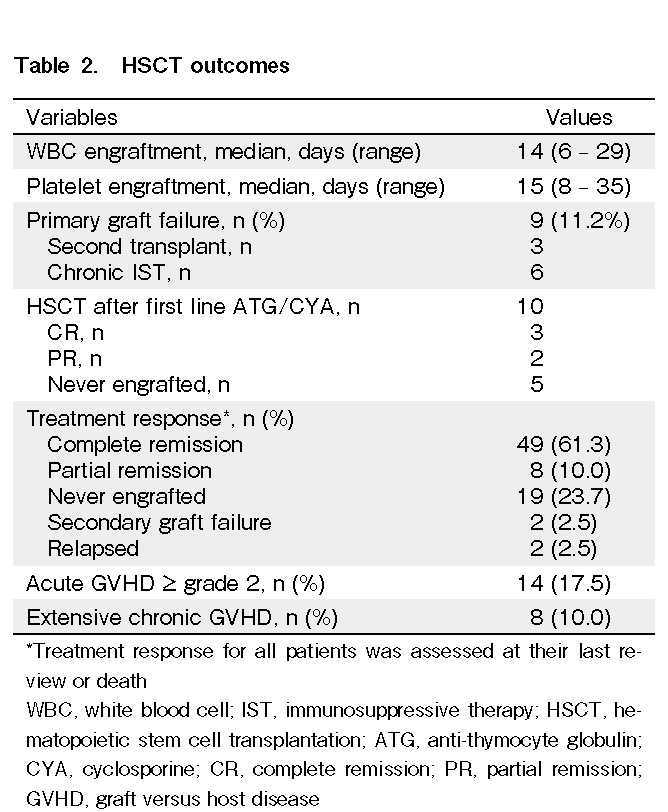
At a median follow-up of 54 months (mean 61, range 0-244 months), 49 patients (61.3%) achieved complete remission (CR) after HSCT, and another 8 patients (10.0%) achieved partial remission (PR). Half of the patients who had prior ATG/CYA as first-line therapy achieved either CR (n = 3) or PR (n = 2). Among the non-responders, none survived even after the second allogeneic transplantation (Table 2). Primary graft failure (GF) was observed in nine patients (11.2%). Of these, only one patient received a second allogeneic transplantation with the same donor who achieved CR. Another two patients who opted for a chronic IST treatment approach achieved PR and were transfusion-independent.
Graft versus host disease
Twenty-three patients developed GVHD following hematopoietic stem cell transplantation. Of these, 17 patients (21.3%) had documented aGVHD, and 14 (17.5%) had ≥ grade 2 aGVHD. The most common sites of aGVHD involvement were the skin (n = 10), gut (n = 7), and liver (n = 3). Thirteen patients (16.3%) developed cGVHD, and eight of these (10%) survived beyond 100 days and had extensive cGVHD. There was no significant difference in the overall incidence of GVHD between BM and PBSCs as stem cell sources. Gender and ABO mismatch, CMV status of patients/donors, and number of red cell transfusions did not significantly impact the incidence of GVHD. A history of preceding aGVHD did not influence the incidence of cGVHD among the patients (Table 4).

Other transplant-related complications
In our cohort of patients, we did not observe any cases of hemorrhagic cystitis or veno-occlusive disease as complications following transplantation. Ninety percent of the patients had febrile neutropenia, and 25% experienced severe sepsis. Grade 1 mucositis was present in 47.5% of the patients, and most of them did not require any parenteral nutrition. Hemorrhagic complications were observed in 11.3% of the patients, and most of them had massive intracranial bleeding. No secondary malignancy developed in any patient, other than a case of post-transplant lymphoproliferative disorder (PTLD). Ninety-five percent of the patients and donors were CMV positive. Of the 11 patients (13.8%) who had CMV reactivation, one of the patients, who was CMV negative, had CMV reactivation from a seropositive donor (Table 5).

Overall survival, mortality, and cause of death
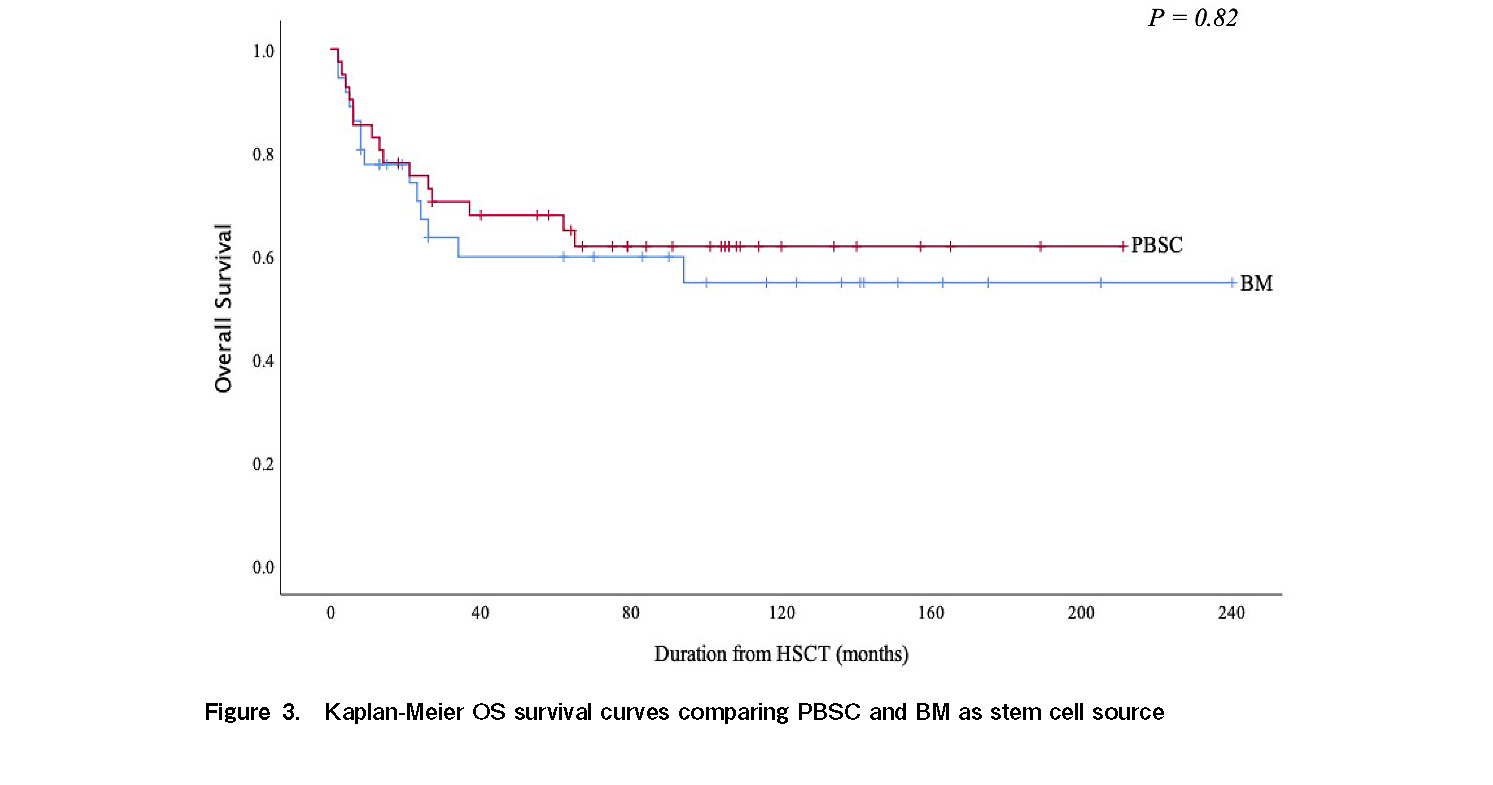
Currently, 50 patients are alive and within the follow-up period. The estimated 5 year OS in our study cohort was 63% (Figure 1). When the study population was stratified into early (transplanted within 3 months from diagnosis) and late (transplanted after 3 months from diagnosis) HSCT, 5 year OS was better (76%) in the former, which reduced to 58% for late transplant (Figure 2). There was no difference in OS among patients who had PBSC as allograft as compared to those who had BM allograft (Figure 3).


Most of the mortality occurred before day +100 (transplant-related mortality, TRM < 100 days, 27.5%). Nearly 50% of the cases had pre-engraftment death, mostly due to infections and bleeding complications. Two deaths occurred due to secondary graft failures. Grade IV steroid-refractory gut aGVHD was the cause of death in two patients, extensive cGVHD with lung involvement in another two patients, three deaths occurred due to poor graft function, and another three patients died from graft rejection.
Discussion
This is the first retrospective study examining the outcomes of all patients with SAA who were referred for HSCT in a tertiary hospital with transplantation facilities. This is an important observational study that evaluated the effectiveness of HSCT treatment among local patients with SAA.
Based on our observations, most of the cases referred for allogeneic transplantation in our center were in the adolescent age group. Our local practice for transplantation among patients with SAA follows the standard guidelines14, 15. MSD transplant is the preferred choice of treatment for SAA patients below 40 years of age. It was found that there was a very strong age effect in patients grafted from matched siblings, with survival rates of 82%, 72%, and 53% for patients aged 1-20, 21-40, and > 40 years, respectively, as a result of the higher incidence of GF and GVHD17. Over the years, there has been significant progress with unrelated donor transplants among SAA patients, and their survival is now comparable to that of sibling grafts18. In our study cohort, only one patient had an unrelated donor transplant after failing first-line IST with ATG/CYA. The patient achieved PR and died from massive intracranial bleeding before day +100. The delay in transplantation and decision for first line ATG/CYA for this patient was probably due to the unavailability of a MSD, a small pool of local MUD, and the process of acquiring an international donor.
Malaysia is a multiracial country that consists of Malay, Chinese, Indian, and other ethnic groups that are unique to Sabah and Sarawak, and some parts of Peninsular Malaysia. Malays are the predominant ethnic group in Peninsular Malaysia and constitute 63.1% of the population. The Ibans constitute 30.3% of the total citizens in Sarawak, while the Kadazan/Dusun constitute 24.5% of the population in Sabah19. Hence, it is not unexpected that Malays and Selangor states have the highest number of SAA cases in Malaysia. In Sabah, the incidence of confirmed AA cases of 4.8 per million is higher than that reported elsewhere in the South East Asian region4. The majority of these people were rural dwellers with an agricultural background and had a lower socioeconomic status. The higher incidence among the Kadazan/Dusun ethnic group suggests a common ethnic environmental factor or genetic predisposition for AA. Environmental causes include traditional treatments, agricultural toxins, and soil-related marrow-suppressive agents6. Poor economic status and educational background probably influence the treatment option among Sabahans as they need to be referred to a transplantation center, which requires them to travel away from home for a minimum of three months.
The current analysis in our study suggests that transplantation within 3 months from diagnosis results in higher survival compared to transplantation occurring after 3 months from diagnosis. This is similar to the observations of a previous study17. For the SAA cases referred from East Malaysia, in particular Sabah, delay in transplantation may be attributed to poor insight, delay in obtaining HLA reports (potential donors may not be easily contactable due to geographical factors), and the need for special arrangements, as most patients had poor education and social background. Nevertheless, delaying transplantation beyond 3 months from diagnosis had an adverse effect on survival, and efforts to limit delays should be encouraged. Setting up an additional transplantation center in East Malaysia to overcome patients’ logistic issues may be ideal for improving outcomes among SAA cases. The delay between diagnosis and transplantation is a modifiable factor, and if transplantation is being considered, our results support an early referral.
The majority of our study patients required red cell transfusion prior to transplantation, and most of them were heavily transfused, possibly due to delays in transplantation. In our local practice, infusions of deferoxamine, at least 7 d prior to conditioning, are administered to patients who received multiple transfusions. Flu-Cy-ATG was selected as the conditioning regimen for patients with multiple transfusions. We did not find a correlation between hematopoietic recovery, increased incidence of GVHD, and adverse outcomes in patients who received > 10 red cell transfusions prior to transplantation in our study population. However, Sakura et al. reported that allogeneic HSCT recipients who received more red cell transfusions during peri-transplantation had an increased risk of developing severe GVHD and had lower OS20. Several other studies have reported inferior OS among patients with elevated pre-transplant ferritin levels. This was not observed in our study cohort, likely due to our small sample size21–23. In our study, we did not examine platelet transfusion or other blood product requirements before or after transplantation. Further studies are required to examine this association with treatment outcomes in SAA patients receiving HSCT.
Multiple studies have reported that the use of BM results in better survival and a reduced risk of severe GVHD compared to PBSCT24–26. In our local practice, we do not perform a red cell-depleted marrow allograft because of cost restrictions. Patients with minor mismatched or matched ABO donors receive BM allografts. PBSC would be an alternative choice when BM harvest is not feasible due to various factors such as donor refusal, limitations in manpower, and operation theatre availability. This could explain the higher usage of PBSC as a stem cell source in our study cohort. Although further evaluation is needed, this may also explain the higher overall survival in the cohort that used the PBSC graft. With regard to hematopoietic recovery and risk of GVHD, we did not observe early engraftment and increased risk of GVHD in the patient who received PBSC, possibly due to the use of G-CSF-primed BM in our center.
More than half of the transplanted patients underwent IST prior to transplantation. Most patients received IST as bridging therapy while waiting for transplantation. Only a small number of patients received ATG/CYA as first-line therapy. From our observation, patients who received first-line therapy with ATG/CYA had poorer outcomes following allogeneic HSCT. Although treating a patient with transplantation alone in the case of IST failure is an optimal strategy, outcomes in patients undergoing transplantation after failing IST are worse than those in patients treated with first-line MSD allogeneic HSCT27. We also found that the outcome of a second allogeneic transplant for patients with primary graft failure was not encouraging. A study on second allogeneic transplantation in children with SAA by Kudo et al. showed favorable results with high OS and failure-free survival (FFS)28. This suggests that a second allogeneic transplant is an option for salvage therapy in treating graft failure29. Our results differ from those of their study, probably due to the small sample size and the inclusion of different subsets of the population.
In this retrospective study, we found that the 5 year OS among patients with SAA who underwent allogeneic HSCT was 63%. Our results appear to be lower than those of other studies30,31. There were more patients who received transplantation after 3 months in our study cohort. This could explain the difference in the OS of our center with those of other studies. Pre-engraftment death was the major cause of TRM < 100 days in our study cohort. We did not observe any trend towards PBSC or BM with pre-engraftment failure, which led to death. As SAA is a non-malignant hematological disorder, avoiding GVHD is a major challenge in HSCT. We found two patients who died from severe steroid-refractory gut aGVHD, and another two patients who died from extensive cGVHD with lung involvement. From our observation, both patients who died due to gut aGVHD were heavily transfused and iron overloaded. On the other hand, both patients received PBSC grafts, and one of them had > 10 red cell transfusions for transplantation. Based on this observation, we suggest that transfusion history and stem cell source play an important role in determining the risk of GVHD and long-term outcomes among patients with SAA who received allogeneic transplantation.
The limitations of this study are that a limited number of patients were included, and it was a retrospective study where not all data were available for analysis. Although the analysis was conducted at a major transplantation referral center in Malaysia, a comprehensive multicenter prospective study of the entire nation will be beneficial. Nonetheless, our study provides an overview of SAA in this region and highlights the possible factors affecting the transplantation outcome in this group of patients.
In conclusion, an early transplantation strategy is necessary to improve outcomes in patients with SAA. Further study is required to look at the modifiable factors leading to delays in referral to the transplant center. Strategies to optimize treatment-related complications are crucial in order to minimize morbidity and mortality among transplanted patients and improve transplantation outcomes among them.
Acknowledgments
The authors would like to thank the Director-General of Health Malaysia for his permission to publish this article.
Author Contributions
W.G, E.S.C, K. W. H and S. A. K. S. S performed the study design. W.G wrote the manuscript and review of the literature. W.G, K. W. H, S. A. K. S. S, T. C. O, and S. M. T contributed information and data for manuscript writing. W.G and E.S.C performed the data analysis. T. C. O, S. M. T, S. A. K. S. S and J. S supervised the manuscript writing.
Acknowledgments
The authors would like to thank the Director-General of Health Malaysia for his permission to publish this article.
Ethical Approval
This study was approved by the Institutional Review Board (IRB) of Medical Research and Ethics Committee (MREC), Ministry of Health Malaysia [NMRR ID-21-02177-UZC].
Conflicts of Interest
The authors declare no conflict of interest. Disclosure forms provided by the authors are available on the website.
References
1.Montané E, Ibáñez L, Vidal X, Ballarín E, Puig R, García N, et al. Epidemiology of aplastic anaemia: a prospective multicenter study. Haematologica. 2008; 93: 518-23.
2.Camitta BM, Thomas ED, Nathan DG, Santos G, Gordon-Smith EC, Gale RP, et al. Severe aplastic anaemia: a prospective study of the effect of early marrow transplantation on acute mortality. Blood. 1976; 48: 63-70.
3.Issaragrisil S, Kaufman DW, Anderson T, Chansung K, Leaverton PE, Shapiro S, et al. The epidemiology of aplastic anaemia in Thailand. Blood. 2006; 107: 1299.
4.Levy M, Kelly JP, Kaufman DW, Shapiro S. Risk of agranulocytosis and aplastic anaemia in relation to history of infectious mononucleosis: a report from the international agranulocytosis and aplastic anaemia study. Ann Hematol. 1993; 67: 187.
5.Young NS, Kaufman DW. The epidemiology of acquired aplastic anaemia. Haematologica. 2008; 93: 489.
6.Yong AS, Goh AS, Rahman M, Menon J, Purushothaman V. Epidemiology of aplastic anaemia in the state of Sabah. Med J Malaysia. 1998; 53: 59-62.
7.Li SS, Hsu YT, Chang C, Lee SC, Yen CC, Cheng CN, et al. Incidence and treatment outcome of aplastic anaemia in Taiwan-real-world data from single-institute experience and a nationwide population-based database. Ann Hematol. 2019; 98: 29-39.
8.Nakagawa MM, Rathinam CV. Constitutive activation of the canonical NF-kappa B pathway leads to bone marrow failure and induction of erythroid signature in haematopoietic stem cells. Cell Rep. 2018; 25: 2094-109.
9.Maciejewski J, Selleri C, Anderson S, Young NS. Fas antigen expression on CD34+ human marrow cells is induced by interferon gamma and tumour necrosis factor alpha and potentiates cytokine-mediated haematopoietic suppression in vitro. Blood. 1995; 85: 3183-90.
10.Risitano AM, Maciejewski JP, Green S, Plasilova M, Zeng W, Young NS. In-vivo dominant immune responses in aplastic anaemia: molecular tracking of putatively pathogenetic T-cell clones by TCR beta-CDR3 sequencing. Lancet. 2004; 364: 355-64.
11.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anaemia. Blood. 2006; 108: 2509-19.
12.Marsh JC, Ball SE, Darbyshire P, Gordon-Smith EC, Keidan AJ, Martin A, et al. Guidelines for the diagnosis and management of acquired aplastic anaemia. Br J Haematol. 2003; 123: 782-801.
13.Scheinberg P. Aplastic Anaemia: therapeutic updates in immunosuppression and transplantation. Hematology Am Soc Hematol Educ Program. 2012; 292-300.
14.Bacigalupo A. How I treat acquired aplastic anaemia. Blood. 2017; 129: 1428-36.
15.Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2015; 172: 187-207.
16.Schoemans HM, Lee SJ, Ferrara JL, Wolff D, Levine JE, Schultz KR, et al. EBMT-NIH-CIBMTR task force position statement on standardized terminology and guidance for graft versus host disease assessment. Bone Marrow Transplant. 2018; 53: 1401-15.
17.Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anaemia using HLA-matched sibling donors. Haematologica. 2010; 95: 2119-25.
18.Bacigalupo A, Socié G, Hamladji RM, Aljurf M, Maschan A, Kyrcz-Krzemien S, et al. Aplastic Anaemia Working Party of the European Group for Blood Marrow Transplantation. Current outcome of HLA identical sibling versus unrelated donor transplants in severe aplastic anaemia: an EBMT analysis. Haematologica. 2015; 100: 696-702.
19.Population distribution and basic demographic characteristics report: Population and Housing census 2010 (Updated: 05/08/2011),
20.Hosoba S, Waller EK, Shenvi N, Graiser M, Easley KA, Al-Kadhimi Z, et al. Peritransplantation red blood cell transfusion is associated with increased risk of graft-versus-host disease after allogeneic haematopoietic stem cell transplantation. Biol Blood Marrow Transplant. 2018; 24: 973-82.
21.Armand P, Kim HT, Cutler CS, Ho VT, Koreth J, Alyea EP, et al. Prognostic impact of elevated pre transplantation serum ferritin in patients undergoing myeloablative stem cell transplantation. Blood. 2007; 109: 4586-8.
22.Lim ZY, Fiaccadori V, Gandhi S, Hayden J, Kenyon M, Ireland R, et al. Impact of pre-transplant serum ferritin on outcomes of patients with myelodysplastic syndromes or secondary acute myeloid leukaemia receiving reduced intensity conditioning allogeneic haematopoietic stem cell transplantation. Leuk Res. 2010; 34: 723-7.
23.Mahindra A, Bolwell B, Sobecks R, Rybicki L, Pohlman B, Dean R, et al. Elevated pretransplant ferritin is associated with a lower incidence of chronic graft-versus-host disease and inferior survival after myeloablative allogeneic haematopoietic stem cell transplantation. Br J Haematol. 2009; 146: 310-6.
24.Schrezenmeier H, Passweg JR, Marsh JC, Bacigalupo A, Bredeson CN, Bullorsky E, et al. Worse outcomes and more chronic GVHD with peripheral blood progenitor cells than bone marrow in HLA-matched sibling donor transplants for young patients with severe acquired aplastic anaemia. Blood. 2007; 110: 1397-400.
25.Bacigalupo A, Socié G, Schrezenmeier H, Tichelli A, Locasciulli A, Fuehrer M, et al. Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anaemia: survival advantage for bone marrow in all age groups. Haematologica. 2012; 97: 1142-8.
26.Chu R, Brazauskas R, Kan F, Bashey A, Bredeson C, Camitta B, et al. Comparison of outcomes after transplantation of G-CSF stimulated bone marrow grafts versus bone marrow or peripheral blood grafts from HLA-matched sibling donors for patients with severe aplastic anaemia. Biol Blood Marrow Transplant. 2011; 17: 1018-24.
27.Ades L, Mary JY, Robin M, Ferry C, Porcher R, Esperou H, et al. Long-term outcome after bone marrow transplantation for severe aplastic anaemia. Blood. 2004; 103: 2490-7.
28.Kudo K, Muramatsu H, Yoshida N, Kobayashi R, Yabe H, Tabuchi K, et al. Second allogeneic haematopoietic stem cell transplantation in children with severe aplastic anaemia. Bone Marrow Transplant. 2015; 50: 1312-5.
29.Cesaro S, Peffault de Latour R, Tridello G, Pillon M, Carlson K, Fagioli F, et al. Second allogeneic stem cell transplant for aplastic anaemia: a retrospective study by the severe aplastic anaemia working party of the European Society for blood and bone marrow transplantation. Br J Haematol. 2015; 171: 606-14.
30.Locasciulli A, Oneto R, Bacigalupo A, Socié G, Korthof E, Bekassy A, et al. Outcome of patients with acquired aplastic anaemia given first line bone marrow transplantation or immunosuppressive treatment in the last decade: a report from the European Group for Blood and Marrow Transplantation (EBMT). Haematologica. 2007; 92: 11-8.
31.Dufour C, Pillon M, Passweg J, Socié G, Bacigalupo A, Franceschetto G, et al. Outcome of aplastic anaemia in adolescence: a survey of the severe aplastic anaemia working party of the European Group for Blood and Marrow Transplantation. Bone Marrow Failure. Haematologica. 2014; 99: 1574-81.
Search
News


