Volume 5 (2022) Issue 1 No.1 Pages 1-15
Abstract
Haematopoietic stem cell transplants (HSCT) are used in the treatment of blood cancers, autoimmune diseases, and metabolic disorders. Over 1.5 million transplants have been performed around the world thus far. In an attempt to enhance the efficacy of the cells used for transplantation, efforts are underway to use cellular engineering to increase cell numbers through: (1) the expansion of hematopoietic stem and progenitor cells (HSPC); (2) cellular subset selection to remove cells that cause graft-versus-host disease (GvHD), while adding back cells, which can mediate anti-tumor and anti-viral immunity; (3) the use of immune regulatory cells, such as mesenchymal stromal cells (MSC) and regulatory T cells (Tregs) to control GvHD; (4) the use of immune effector cells to mount immunological control of tumor cells before, after, or independent of blood stem cell transplants.
Introduction
Haematopoietic stem cell transplants (HSCT) are used in the treatment of blood cancers, autoimmune diseases, and metabolic disorders1–3, with over 1.5 million transplants performed thus far around the world and over 50,000 performed annually4. Cells used for HSCT are obtained from bone marrow (BM), peripheral blood (PB), or umbilical cord blood (UCB). There are two types of HSCTs: (1) autologous HSCTs, wherein hematopoietic stem and progenitor cells (HSPC) are taken from the patients themselves; (2) allogeneic HSCTs, wherein the cells are taken from a donor5. However, allogeneic HSCT performed using blood cells from a donor could lead to graft-versus-host disease (GvHD), a potentially fatal condition in which donor immune cells attack the recipient's body5. The lack of suitable human leukocyte antigen (HLA)-matched donors has been overcome by using haploidentical and UCB donors. Haploidentical (“half-matched”) family donor transplants have been successful through the use of in vitro and in vivo T cell depletion methods, which reduce donor T cells and GvHD. UCB transplants have significant benefits due to their ability to perform HLA-mismatched transplants without in vivo or in vitro T cell depletion. Furthermore, UCB grafts retain a strong graft versus tumor (GVT) effect while having a lower incidence of GvHD compared to the other two sources of HSPCs7. However, UCB contains limited numbers of HSPCs, which has led to delayed engraftment and immune reconstitution, as well as increased rate of graft failure6–8. Currently, the main challenge associated with HSCTs lies in the delivery of an adequate number of cells to enable rapid hematological reconstitution while minimizing GvHD and maximizing GVT of infused donor cells. In this review, we discuss methods to improve the outcome of patients, including cell selection in haploidentical transplantations, the use of immune regulatory cells including mesenchymal stromal cells (MSCs) and regulatory T (Treg) cells, HSPC expansion, and immune effector cell therapy using donor lymphocyte infusions (DLI), chimeric antigen receptor (CAR) T cells, natural killer (NK) cells, and gamma delta (γδ) T cells (Figure 1).
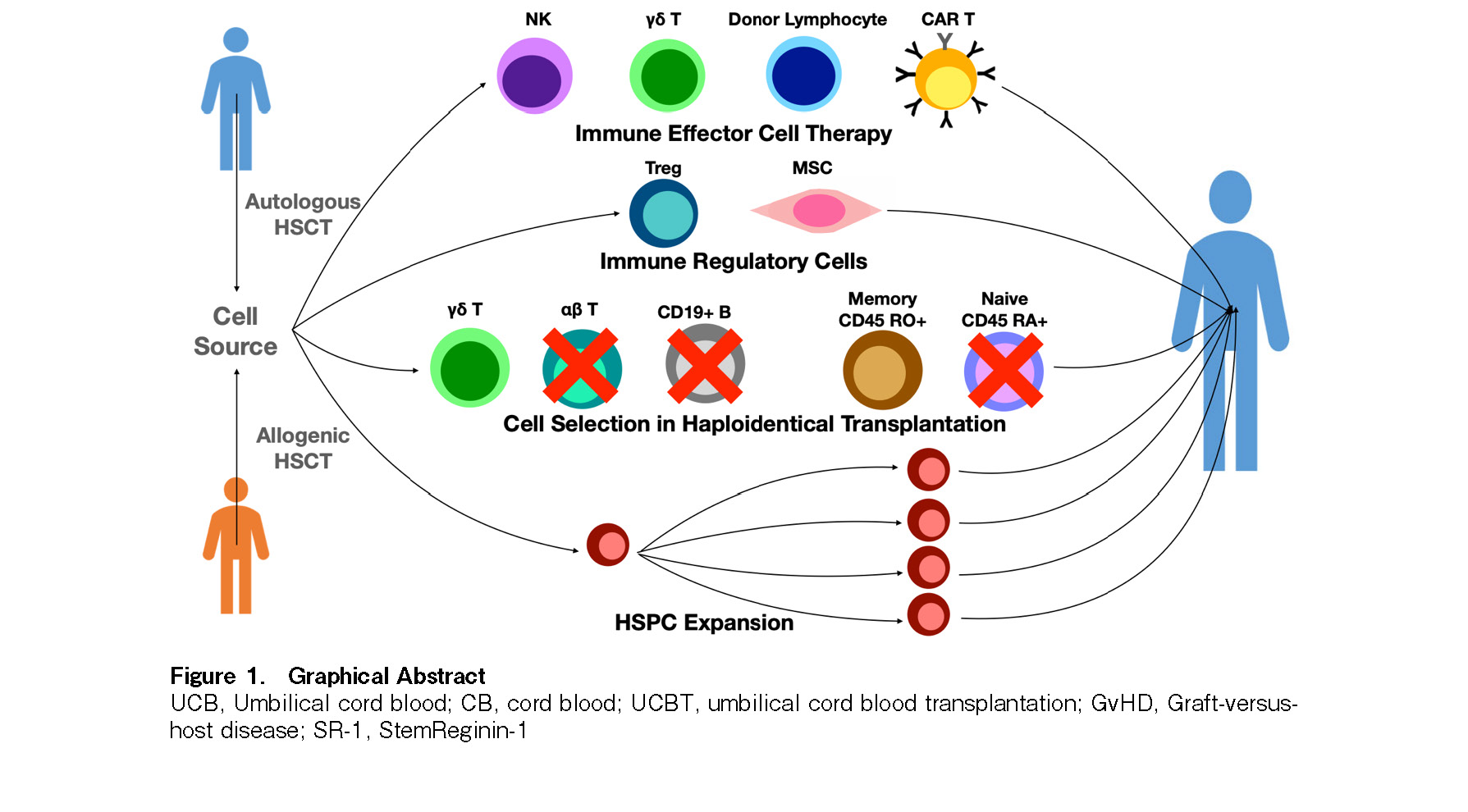
HSPC Expansion
Umbilical cord blood expansion (Table 1)
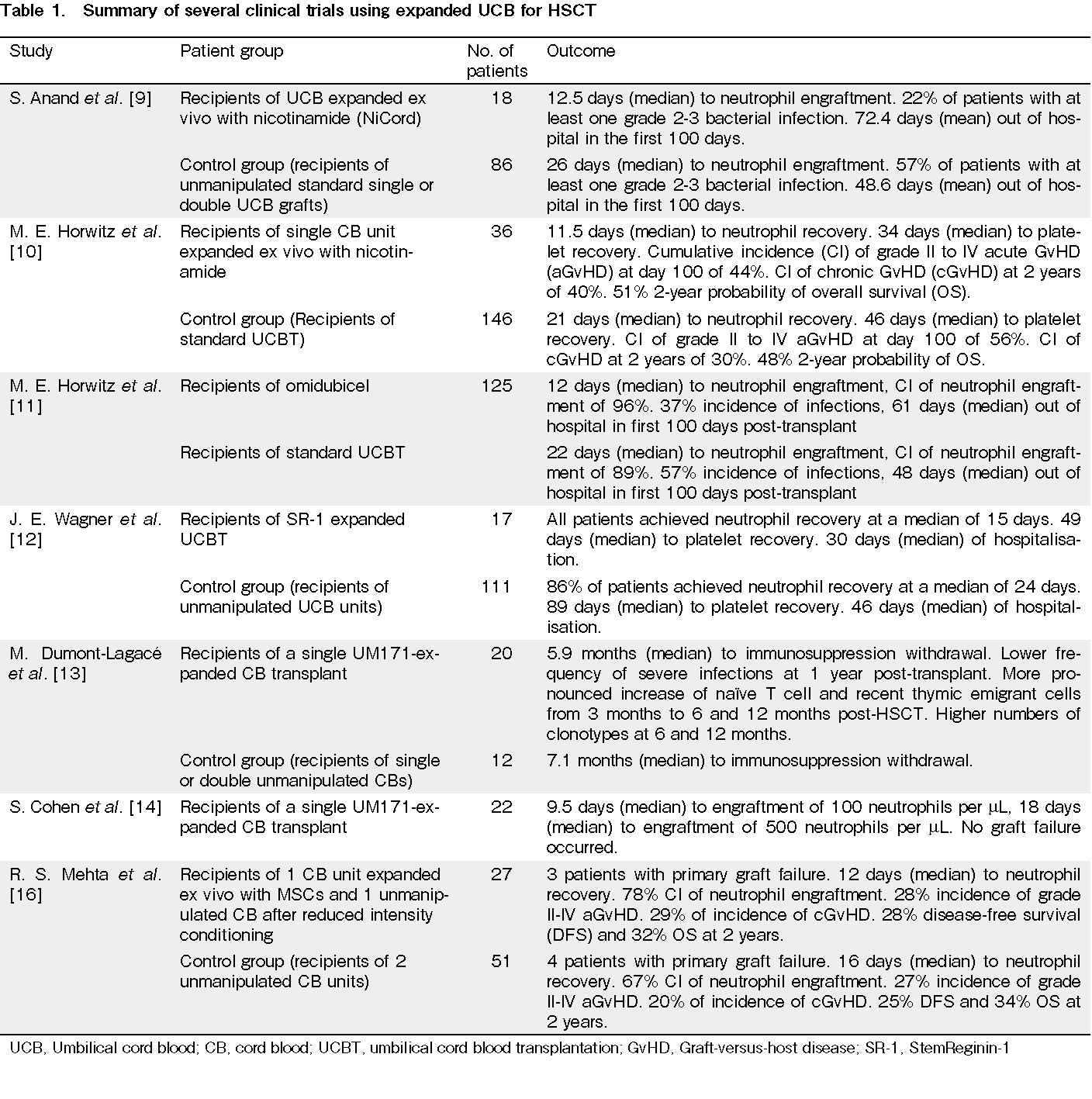
To optimize the outcomes of patients undergoing HSCTs performed using UCB, HSPCs must undergo expansion in order to increase the number of HSPCs, thereby decreasing graft failure rates and increasing survival rates. Several studies have investigated the use of different cultures for UCB expansion. Clinical trials involving transplantation have used NiCord, an ex vivo expanded cell product derived from UCB, which uses nicotinamide as the active agent to inhibit differentiation and enhance the functionality of cultured HSPCs9, 10. These trials have shown the multi-fold expansion of CD34+ cells, earlier median neutrophil recovery, and long-term engraftment, with no adverse outcomes associated with the use of NiCord9, 10. In a recent study, 36 patients underwent transplantation with NiCord as a stand-alone graft between 2013 and 2017. After NiCord expansion, a 33-fold increase in the CD34 content of the graft was observed, and by 21 days after transplantation, 89% of NiCord recipients had achieved neutrophil engraftment. Furthermore, neutrophil engraftment was faster for the recipients of NiCord. NiCord recipients also had a higher incidence of platelet engraftment (81%) 100 days after HSCT, with faster platelet engraftment and recovery10. In another study conducted with omidubicel, formerly known as NiCord, 125 patients with hematologic malignancies received either omidubicel or standard umbilical cord blood transplantation (UCBT)11. The results showed that the omidubicel transplant recipients had a shorter median time to neutrophil engraftment of 12 days compared to 22 days for patients in the control group. Omidubicel recipients also had a higher cumulative incidence (CI) of neutrophil engraftment, as well as a faster platelet recovery, a lower incidence of bacterial and fungal infections, and they spent more time out of hospital during the first 100 days post-transplant as compared to the control group11.
In addition to NiCord, StemReginin-1 (SR-1) is an aryl hydrocarbon receptor antagonist that has also been studied for its effect on UCB expansion and HSCT12. In a phase I/II trial, 17 patients received HSC835, which was the product of the expansion of UCB CD34+ cells using SR-112. After 15 days in SR-1 expansion culture, the median number of total CD34+ cells was found to increase 330-fold from an initial 4.4 × 106 cells to a final 1,440 × 106 cells. Patients who had received HSC835 all demonstrated neutrophil recovery, which was achieved faster than the control group12. Another expansion technique involves the use of UM171, an HSPC agonist, which has been shown in several studies to successfully expand UCB units13, 14. In one of the studies, 20 patients received a single UM171-expanded UCB transplant, while 12 patients received transplants with single or double unmanipulated UCB. The results showed a 17-fold larger CD34+ cell dose and a 2-fold lower CD3+ T cell dose in UM171 patients, as well as a shorter median time to immunosuppression withdrawal for the UM171 cohort, compared to the control cohort13. A novel azole small molecule, C7, was used in the ex vivo expansion of UCB HSPC, and the results showed that the addition of C7 could boost the expansion of hematopoietic progenitor cells (HPCs) by large amounts. The culturing of UCB CD34+ cells in the presence of C7 resulted in a 283.7 ± 14.7‐fold increase of viable CD45+CD34+CD38–CD45RA– HPCs within 11 days15. The limitations of UCB in terms of its low blood cell count, which prevented the achievement of the most optimal outcomes in patients post-HSCT, can now be overcome by HSPC expansion through culturing with various substances. In fact, several studies have reported large increases in the number of HSPCs, and subsequent improved outcomes in patients.
Other indications for HSPC expansion
Besides the expansion of HSPCs from UCB, HSPCs derived from the BM and PB can also be expanded to improve the outcomes of HSCTs. Regardless of the source of HSCs, the success of HSCT is affected by the number of transplanted cells. A sufficiently large number of HSCs must be transplanted for HSCT to succeed17, 18. Thus, HSPC expansion techniques can also be useful when the number of cells obtained from BM or PB stem cell harvest is insufficient for HSCTs, or when insufficient hematopoietic progenitor cells are mobilized into the bloodstream. Hence, there is potential for the expansion of HSPCs to achieve an adequately large number of cells for a successful transplant19, 20. Another possible use for HSPC expansion that is becoming increasingly relevant is in gene therapy, since after the HSCs have been genetically modified, the number of genetically modified cells may be low. Thus HSPC expansion is needed to achieve the minimum cell dose before the transplant can occur21, 22. In addition, HSCT and chemotherapy are often followed by a period of neutropenia in which faster neutrophil recovery results in better outcomes for the patient23. Therefore, HSPC expansion has also been explored as a potential treatment to reduce the period of neutropenia after HSCT and chemotherapy, enhancing neutrophil recovery and reducing the chances of infections10, 12, 23.
Cell Selection
Haploidentical transplantation (Table 2)

To overcome the risk of GvHD due to T cell alloreactivity with haploidentical transplantations, there is a need for in vitro or in vivo T cell depletion. However, these processes could also blunt the effect of GVT. Thus, there is a need for research exploring how to maximize the anti-leukemic effects of the graft while removing the GvHD effect of transplanted cells24. One of the current methods for haploidentical transplantation is TCR αβ+/CD19+ cell depletion, in which there is a selective depletion of T cells expressing the αβ T cell receptor (TCR) and CD19+ B cells before transplantation, as these cells are responsible for GvHD25–27. However, T cells expressing γδ TCR are retained as γδ T cells are able to produce the GVT effect without causing GvHD25, 27. In one study, thirty-seven patients with primary immunodeficiency (PID) received transplants from a matched unrelated donor (MUD) or haploidentical mismatched related donor (MMRD), with all grafts being TCR αβ+/CD19+ depleted28. Patients received a median of 11.7 × 106 /kg CD34+ cells and 10.6 × 103 /kg TCR αβ+ cells. As a result, neutrophil engraftment occurred in 35 of 37 patients 11‒28 days after transplantation, with grade II GvHD occurring in only 7 cases and grade IV GvHD occurring in 1 case, corresponding to a 21.5% probability of grade II GvHD and 2.8% probability of grade IV GvHD28. With TCR αβ+/CD19+ cell depletion, the GvHD probability and severity can be maintained at a moderate level. Together with TCR αβ+/CD19+ cell depletion, another method currently used to lower GvHD rates and increase survival rates in haploidentical transplants involves the depletion of naïve CD45RA+ cells while retaining memory CD45RO+ cells29–31. Naïve CD45RA+ cells are selectively depleted, as they can lead to GvHD, while memory CD45RO+ cells are retained, as they can help to protect the body against infections, thereby maintaining the GVT effect without causing GvHD29–31. In one clinical study, 41 patients received CD3-depleted (CD3-dep recipients) progenitor cell products (HPC,A) and 26 received CD45RA-depleted HPC,A (CD45-dep recipients)31. The results showed faster T cell recovery in CD45RA-dep recipients, with a median T cell count of 550 T cells/μL after 30 days compared to only 10 T cells/μL in the CD3-dep recipients. Furthermore, quantitative B cell recovery occurred earlier in CD45RA-dep recipients, while NK cell recovery was quantitatively higher in CD3-dep recipients31. For CD3-dep recipients, there were two consecutive trials. In trial 1, six out of the ten patients had acute GvHD (aGvHD) grade III-IV, while in trial 2, seven out of 31 patients had aGvHD grade III-IV. Furthermore, for CD45RA-dep recipients, six out of 26 had aGvHD grade III-IV31. The results of this study also showed that 76% of CD3-dep recipients had detectable viremia in the first 180 days post-transplant, compared to only 35% of CD45RAdep-recipients. In addition, 58% of the viremic CD3dep-recipients had prolonged viremia compared to only 11% of the viremic CD45RAdep-recipient31. Thus, both CD45RA-dep recipients and CD3-dep recipients had similar GvHD rates, indicating that both the depletion of CD45RA cells and CD3 cells can maintain GvHD rates at a moderate level. However, CD45RA depletion is the optimal treatment option, as it results in a faster immune reconstitution and allows for the preservation of immunity against viruses, leaving transplant recipients less susceptible to viral infections30, 32.
Immune Regulatory Cells
Mesenchymal stromal cells (MSC) (Table 3)
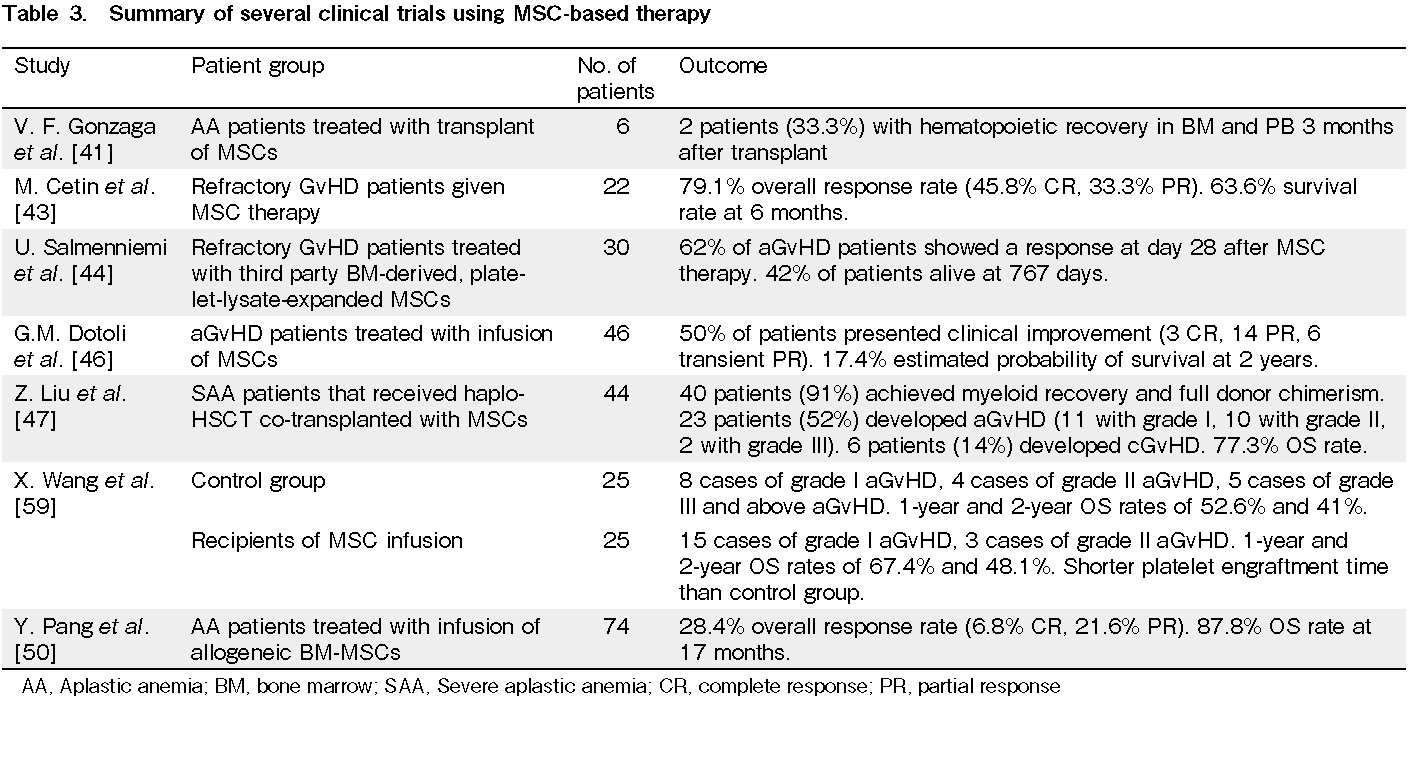
MSCs are multipotent spindle-shaped plastic-adherent cells isolated from the BM, adipose, and other tissue sources37, which can differentiate into cells of several lineages38. Due to the ability of MSCs to interact with both the innate and adaptive immune systems, exhibiting a strong immunosuppressive activity on both immune systems and inducing tolerance, many studies have explored the role of MSCs in tissue repair, immune modulation, and the BM microenvironment, and hence the potential of MSCs in clinical treatment for various conditions38–40. One of the conditions that is frequently studied is aplastic anemia (AA), which is a BM failure disorder characterized by marrow hypoplasia and peripheral pancytopenia, which is the reduction in the number of red blood cells (RBCs), white blood cells (WBCs), and platelets (PLTs)41, 42. In a study using animal models, irradiation was used to induce BM failure in the animals, since HSPCs and committed BM progenitor cells are more sensitive to irradiation41. Mice were administered 1 × 106 lymph node cells to induce acquired AA, mimicking AA in humans41. Subsequently, 1 × 106 to 2.5 × 107 MSCs were transplanted into each mouse, and preclinical studies reported an increase in the levels of WBCs, PLTs, and hemoglobin in PB after MSC transplantation compared to the control group, which did not receive MSC transplantation. The group that received MSC transplantation also showed BM recovery and an increase in the number of BM cells in vivo, as well as an increase in cytokines FLT3LG and TGF-beta 1 secreted by MSCs, which are involved in the proliferation and differentiation of HSPCs41. In a clinical trial, six patients with severe AA (SAA) received an MSC transplant, and two had a hematopoietic recovery in both BM and PB three months after transplantation; however, some patients manifested adverse events during or after MSC infusions41. Thus, while MSCs have great potential in the treatment of AA, more research and studies are needed to develop an MSC-based treatment that can avoid the adverse events in most, if not all, patients receiving MSC transplantation. Other than AA, studies have also shown the ability of MSCs to prevent or treat GvHD associated with HCST43–47. A study of 22 patients with refractory GvHD was conducted, with a median dose of infused MSCs of 1.64 × 106 cells/kg, and a total of 79 MSC infusions administered43. The patients' response to MSC infusions was evaluated in terms of no response (NR), which corresponds to no change in GvHD grade, partial response (PR), which corresponds to at least 1 grade decrease in GvHD compared with the patient's initial condition on day 0, and complete response (CR), which corresponds to an absence of GvHD signs. After MSC therapy, the overall response rate was 79.1% for 19 of the patients, with 11 patients achieving CR and 8 patients achieving PR. Among these, 7 out of the 11 CR patients initially only achieved PR after induction therapy but were able to achieve CR after additional MSC therapy43. In addition, the survival rate in patients at 6 months was significantly higher in the CR and PR groups, with a survival rate of 76.5%, compared to the NR group, which had a much lower survival rate of 20%43. These results indicate the effectiveness of MSC therapy in reducing or completely treating GvHD in a significant number of patients. Additional MSC therapy after an initial induction therapy can further improve the outcomes of patients. Another study assessed the feasibility of an MSC-derived 2-factor combination (2FC) of CXCL5 and anti-CCL24 in place of MSCs in the treatment of GvHD48. In this study, GvHD mouse models were produced via the tail vein injection of human PB mononuclear cells (PBMCs) into NSG mice, followed by Cesium1, 37 irradiation in mice 3 to 4h before PBMC transplantation48. The 2FC treatment was administered to the mice 10, 14, 17, and 21 days post-PBMC transplantation, while the extended treatment with reduced dosage was administered 4 , 7, 11, 18, 21, 25, and 28 days post-transplantation48. The results showed that by day 36, survival in mice with moderate GvHD administered 2FC treatment was higher at 81.0% with mild symptoms compared to 50.0% in the control group administered Dulbecco's Phosphate-Buffered Saline (DPBS) with moderate symptoms48. The immunosuppressive effect of 2FC therapy was similar to that of BM-MSC treatment and superior to the cyclosporine A (CsA) treatment48. For mice with severe GvHD, the mice that received the 2FC treatment had higher survival rates compared to the DPBS control group, while the BM-MSC and CsA treatment did not lead to much improvement, and hence had similar survival rates to the control group and lower survival rates than the 2FC therapy group48. Although further clinical trials are required to assess the feasibility of the 2FC treatment for GvHD in humans, the results of the study show that 2FC has great potential in the treatment of GvHD and could even be more effective than MSC therapy.
Regulatory T (Treg) cells
Treg cells have also been explored as a potential treatment to improve the outcomes of HSCTs by treating or preventing GvHD. However, a significant limitation of Treg cells treatment is that they suppress anti-tumor immunity, thereby inhibiting cancer treatment51–56. Treg cells are a type of CD4+ T cells that express the transcription factor Forkhead box P3 (FoxP3) and are involved in the suppression of immune responses against both self- and non-self – antigens. As a result, they are able to ameliorate GvHD, but also worsen the prognosis of cancer patients53, 56. In one of the studies conducted, Tregs were isolated from a unit of UCB matched to the patient, and a total of 11 patients were treated57. Among these, 2 patients were treated at 3 × 106 Treg/kg, 4 were treated at 10 × 106 Treg/kg, 1 was treated at 30 × 106 Treg/kg, and 4 were treated at the highest achievable dose of 100 × 106 Treg/kg57. In addition, there were a total of 22 control patients57. The results showed that for the 11 patients treated with Tregs, the CI of grade II-IV aGvHD at 100 days was 9%, which was much lower than the control group that had a CI of grade II-IV aGvHD of 45% and grade III-IV of 27%. Furthermore, chronic GvHD (cGvHD) developed in 3 out of the 22 patients in the control group, while none of the Treg recipients developed cGvHD57. Another study harvested intestinal tissue from mice; the intestine is the most common tissue involved in GvHD after HSCT. The results showed that the intestinal injury was significantly attenuated in mice that received CD150+ Treg cells, and the number of infiltrated leukocyte common antigen lymphocytes was reduced significantly compared to the control group52. These results indicate the potential for Treg treatment to be used to prevent the development of GvHD in post-HSCT patients. However, the ability of Tregs to support tumor progression and suppress anti-tumor immunity means that further research is needed to develop a safer Treg treatment51.
Immune Effector Cell Therapy
Donor lymphocyte infusions (DLI) (Table 4)
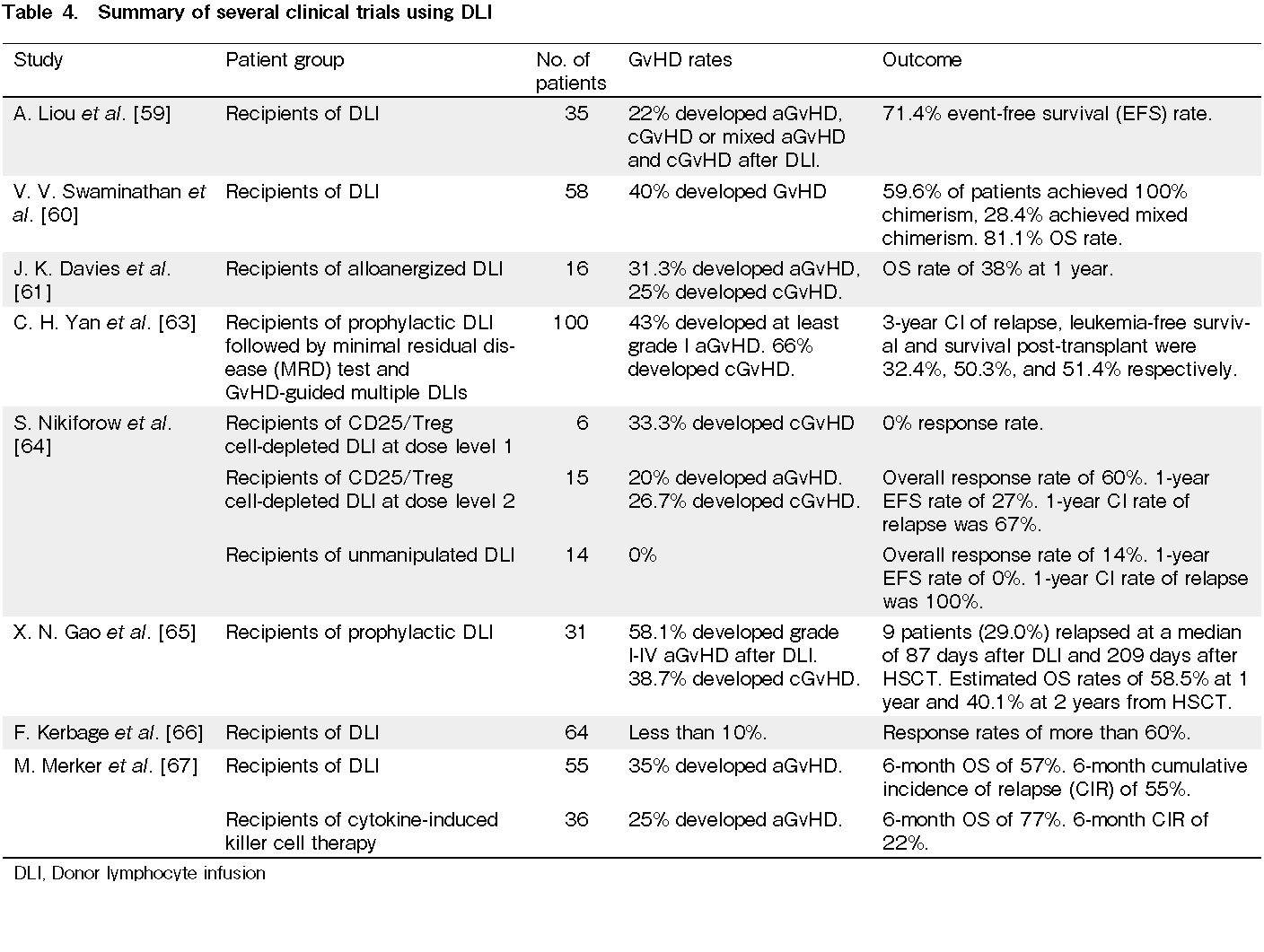
While HSCT remains the only curative treatment for several diseases, such as leukemia, many transplant recipients still do not survive post-transplant due to relapse58, 59. When a patient relapses, one of the treatment methods used is DLI, in which T lymphocytes from the donor are used to induce the GVT effect in patients60. However, while DLI has been shown to produce benefits through the induction of the GVT effect, it often leads to GvHD after infusions61–63. In a study on the safety and effectiveness of DLI in children, DLI was performed in 58 children, of whom 71% had benign hematological conditions and 29% had relapsed or high-risk leukemia/lymphoma60. In this study, 28 children received single DLI, 17 children received two DLI, and 13 children received three DLI60. As a result, 23 children developed GvHD, with an overall survival (OS) rate of 81.1%60. In another study, 21 patients received a CD25/Treg-depleted DLI, a single infusion of fresh cells immediately following selection, resulting in one case of Grade III aGvHD, two cases of grade I aGvHD, and six cases of cGvHD64. Thus, DLI has indeed been associated with higher rates of GvHD. Therefore, there is a need to investigate further methods for the treatment of relapses that are able to induce the GVT effect without causing GvHD.
Chimeric antigen receptor (CAR) T cells (Table 5)
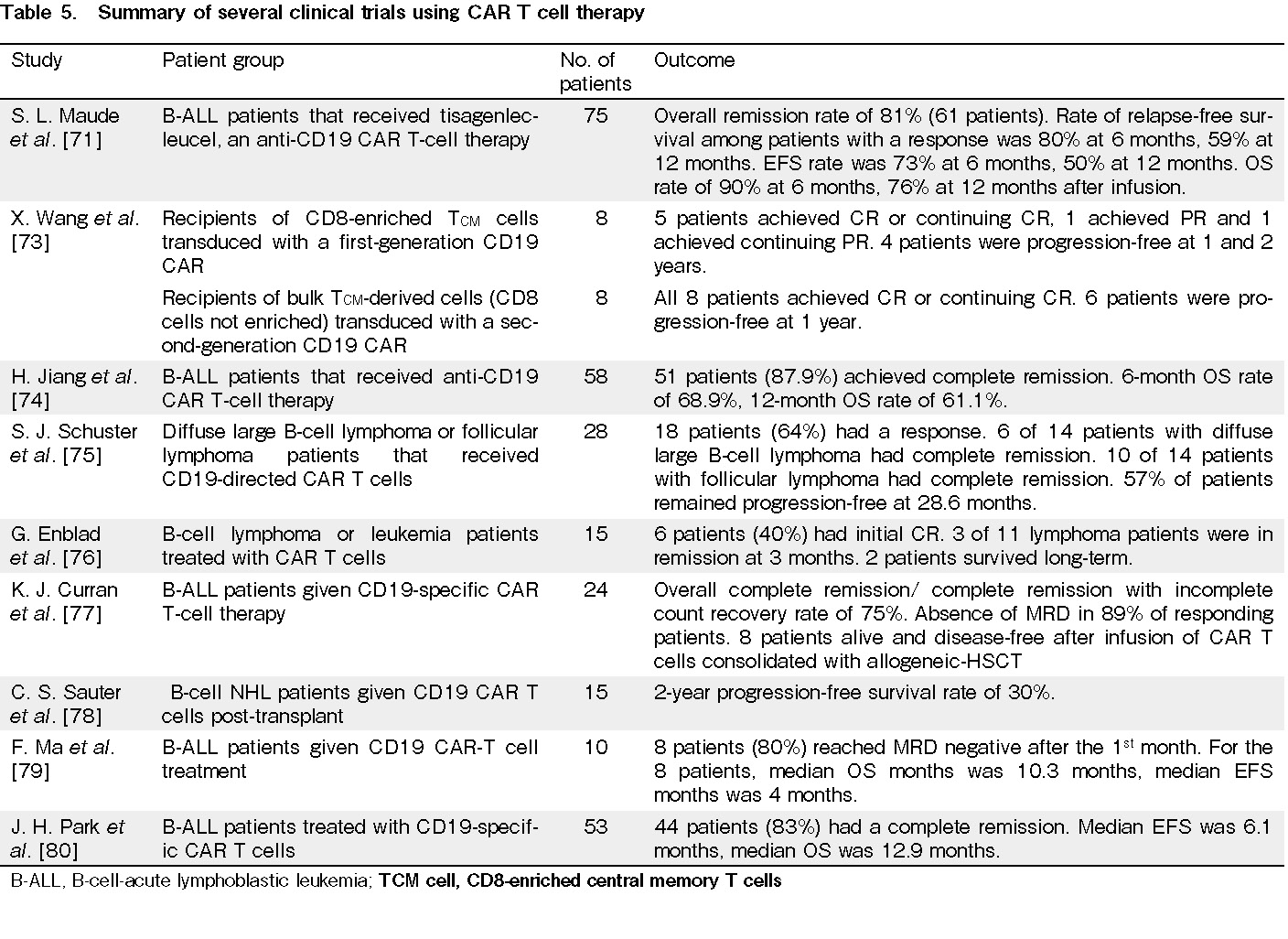
CAR T cells are T cells that are engineered to express CARs, which target the B-cell-specific antigen CD19, as CD19 is expressed at high and stable levels in the tumor tissue of most patients with B-cell-acute lymphoblastic leukemia (B-ALL), non-Hodgkin's lymphoma (NHL), and chronic lymphocytic leukemia (CLL). This allows the CAR T cells to be used in the treatment of these B cell cancers68–70. In a global study on the use of anti-CD19 CAR T cell therapy tisagenlecleucel for B-ALL, 75 patients received tisagenlecleucel71. With at least three months of follow-up, the overall remission rate was 81%, with 45 patients having complete remission and 16 having complete remission with incomplete hematologic recovery; the event-free survival (EFS) rate was 73% at 6 months and 50% at 12 months71. However, all patients with a response to treatment had B-cell aplasia, the median time to B-cell recovery was not reached, and all 75 patients had at least one adverse event during the study, with 95% having an adverse event suspected to be related to tisagenlecleucel71. While CAR T cell therapy has great potential in the treatment of B-ALL and can lead to high remission and survival rates, the high rates of adverse events indicates that further research is needed to develop a CAR T cell therapy that can maintain high remission and survival rates with lower rates of adverse events72. Another study assessed the immunotherapy of NHL in which there were two trials: the first trial (NHL 1) tested CD8-enriched central memory T (TCM) cells transduced with a first-generation CD19 CAR without a costimulatory domain and the second trial (NHL 2) tested bulk TCM-derived cells in which CD8 cells were not enriched, and the bulk TCM cells were transduced with a second-generation CD19 CAR, including a CD28 costimulatory domain73. For NHL 1, 8 patients received HSCT and CD19 CAR TCM cell infusions. As a result, 5 patients achieved CR or continued CR, 1 patient achieved PR, and 1 achieved continued PR73. For NHL 2, 8 patients received HSCT, and all 8 patients achieved CR or continued CR73. While there were high rates of adverse events with all NHL 1 patients and 7 out of the 8 NHL 2 patients experiencing grade 3 non-hematologic toxicities, these were attributed at the probable or definite level to HSCT73. In addition, there were no grade 2 or higher toxicities attributed at the probable or definitive level to CD19 CAR TCM cell infusions, thereby demonstrating the effectiveness of CD19 CAR T cell therapy for NHL73. Furthermore, CAR T cells persist for long periods of time in the blood, further boosting their effectiveness by making them a long-lasting therapy71, 73. Further research should seek to explore whether CAR T cell therapy can be used as an alternative to HSCTs or in combination with transplants. If they are used together with HSCTs, studies should explore whether CAR T cell therapy should be administered to the patient before or after the transplant.
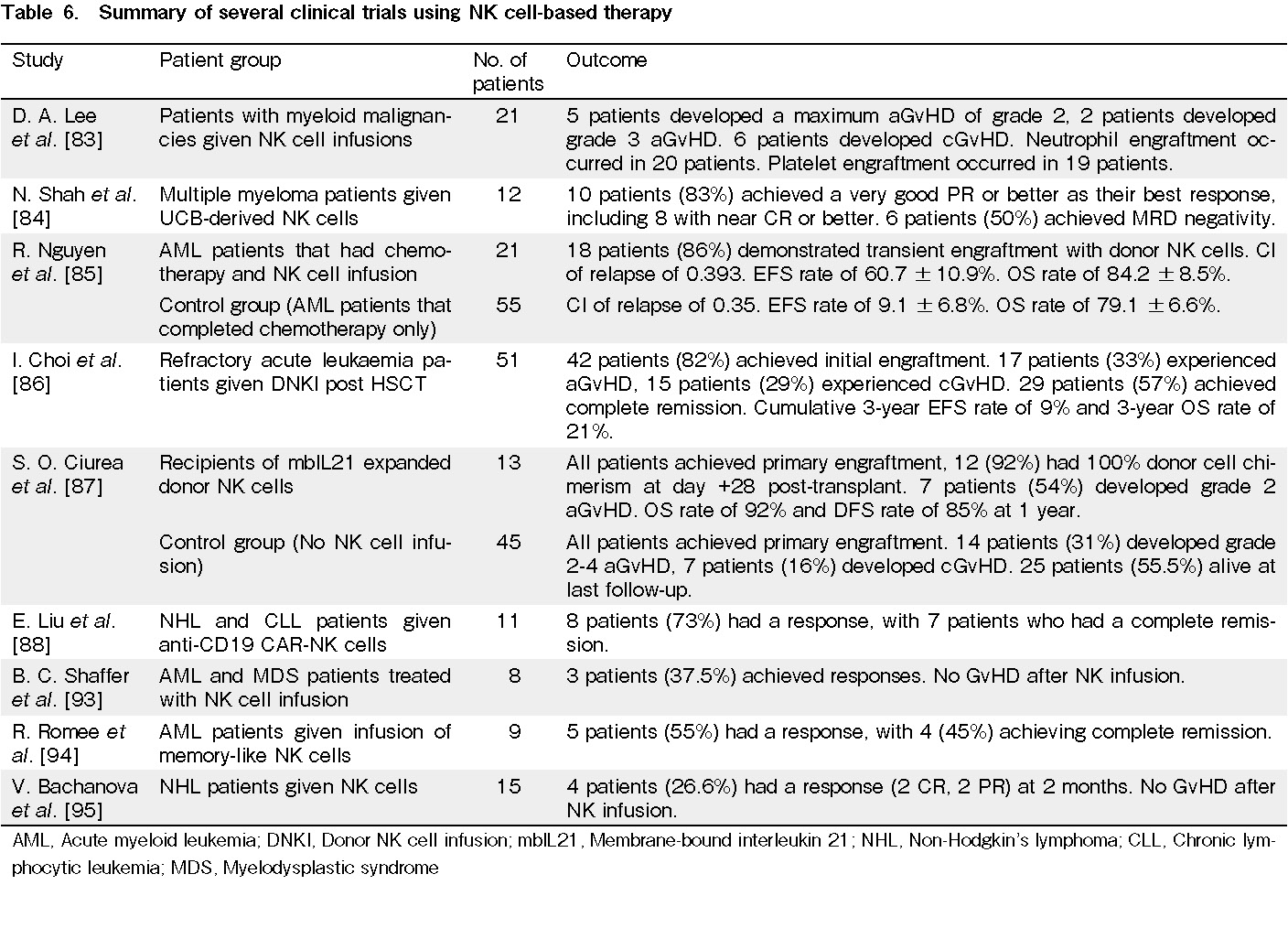
Natural killer (NK) cells
NK cells are part of the innate immune system. They kill virus-infected cells and cancerous cells without requiring prior exposure or sensitization to these cells, thereby allowing them to contribute to the GVT effect. Thus, several studies have explored the use of NK cell-based immunotherapy to improve the outcomes of patients81–84. In a phase II clinical trial, 21 children with acute myeloid leukemia (AML) received NK cell therapy with a median of 12.5 × 106 NK cells/kg85. Although 20 of the 21 patients treated developed grade ≥3 neutropenia, and 5 patients developed grade ≥3 thrombocytopenia, 20 of the 21 patients had absolute neutrophil and platelet count recovery within 45 days after NK cell infusion and none had opportunistic infections, notable bleeding, or GvHD85. However, the results also showed that NK cell infusion did not improve the CI of relapse or OS compared to chemotherapy alone85. In another study, 51 patients with refractory acute leukemia received HSCT followed by at least one donor NK cell infusion (DNKI)86. Among the 51 patients, 24 received all four planned doses of DNKI, while 27 did not receive one or more of the planned doses of DNKI due to cytokine release syndrome, rapid leukemia progression, inadequate NK cell product and CD3+ cells >10%, transient mental change, sepsis, or patient refusal86. The results showed that 42 recipients achieved initial engraftment with an absolute neutrophil counts(ANC) ≥500/μL at a median of 15 days after HSCT, and 18 patients achieved platelet counts ≥20,000/μL at a median of 32 days86. However, 17 patients experienced aGvHD at a median of 1.1 months after HSCT, while 15 patients developed cGvHD at a median of 3.4 months after HSCT86. Of the 51 patients who received at least 1 DNKI, 29 achieved complete remission, and the cumulative 3-year EFS rate was 9%, while the 3-year OS rate was 21%86. In another phase I clinical trial, 13 patients with high-risk AML, myelodysplastic syndrome (MDS), or chronic myeloid leukemia (CML) received membrane-bound interleukin 21 (mbIL21) NK cell infusions87. Primary engraftment was achieved in all 13 patients, with 12 of 13 patients having 100% donor cell chimerism after day 28 post-HSCT87. The median time to neutrophil engraftment was 19 days, while the time to platelet engraftment was 22 days87. However, 7 patients developed grade 2 aGvHD, with no grade 3-4 aGvHD nor cGvHD. Furthermore, the one-year OS rate was 92% while the one-year disease-free survival rate was 85%87. For the control group (45 patients), all patients achieved primary engraftment, with a median time to neutrophil and platelet engraftment of 18 and 40 days. Among these, 14 patients developed grade 2-4 aGvHD, while 7 patients developed cGvHD, with 25 patients alive at the last follow-up87. While NK cell-based immunotherapy is generally safe, toxicities can develop after treatment. However, at present, the effectiveness of NK cell infusions in the treatment of patients is not very significant, and further research is needed to develop a more effective NK cell-based therapy. In a study in which NK cells were engineered to express CAR (CAR-NK cells), 9 patients received a CAR-NK product that was partially matched with the HLA genotype of the recipient, while two patients received a CAR-NK product that did not take HLA matching into consideration88. As a result, at a median follow-up of 13.8 months, 8 patients had an objective response to the treatment that occurred during the first month after infusion88. Seven patients had CR and one patient showed a complete remission of high-grade lymphoma88. With a high response rate and fewer toxicities, this study shows the potential effectiveness and safety of CAR-NK cell infusions in treating patients despite the CAR-NK products being HLA mismatched. Other studies have shown the possibility of producing several doses of CAR NK cells from a single unit of UCB89. Thus, future studies could seek to explore the possibility of making CAR-NK cell infusion a more accessible, universal treatment, since the use of CAR-NK cells could eliminate the need for infused cells to be specific to individual patients. The role of NK cells in cancer treatment has also been explored in anti-tumor monoclonal antibody (mAb) therapies, since one of the mechanisms by which the mAbs target tumor cells is antibody-dependent cell-mediated cytotoxicity (ADCC), which is mediated by NK cells90, 91. The binding of IgG antibodies to the target tumor cells allows for the recruitment of NK cells to the tumor cells via the binding of NK cells' fragment crystallizable receptor (FcR), CD16A, to IgG, thus aiding the NK cells in mediating ADCC to kill tumour cells90, 91. One of the most widely used mAbs, which was also the first mAb therapy used for cancer treatment, is rituximab, which is an IgG1 mAb that targets CD20, a B cell differentiation antigen90, 91. In a study conducted using rituximab, 101 patients with follicular lymphoma were treated with rituximab-containing antibody combinations92. Among these, 46 patients were administered rituximab with galiximab and 55 were administered rituximab with epratuzumab92. Unlicensed NK cells lack the expression of an inhibitory killer immunoglobulin-like receptor (KIR) for self-HLA class I ligands, and are less responsive than licensed NK cells in steady state; however, they can elicit strong responses in inflammatory conditions92. The results of this study showed that coating tumor cells with rituximab allows for the hypo-responsive unlicensed NK cells to be activated and to mediate increased cytotoxicity, contributing to a greater response against the tumor cells by the NK cell repertoire92. Thus, there is great potential for the use of anti-tumor mAb therapies to enhance ADCC by NK cells, and future research should explore other methods of boosting NK cells' responsiveness to tumor cells and mediation of ADCC (Table 6).
Gamma delta (γδ) T cells
There is an increasing amount of research on the potential use of γδ T cells in the treatment of cancer. These cells are not restricted by the need for antigen presentation by major histocompatibility complex (MHC) molecules and are able to produce abundant cytokines, playing a part in both innate and adaptive immunity against tumours96–98. In a study using mice, nude mice were injected intravenously with Daudi cells from the Daudi human tumor cell line99. Three days later, the mice were injected with γδ T cells plus rhIL-2, rhIL-2, or phosphate-buffered saline (PBS). Then, IL-2 was administered on the day of cell transfer, with treatment continuing twice each day for 4 days99. The results showed that the ex vivo-expanded γδ T cells showed potent cytotoxicity against all of the tested lymphoma cell lines, and the survival rate and change in body weight of mice treated with γδ T cells plus IL-2 was significantly higher than that of the PBS or IL-2-treated group99. Furthermore, no lymphoma cells were found in the BM smears from mice in the γδ T cells plus IL-2 treatment group, while two IL-2-treated mice had 7% and 8% of lymphoma cells in the BM smears, with a percentage that was even higher for two PBS-treated mice (21% and 24%)99. In another study, four patients with advanced refractory hematological malignancies who were not eligible for allogeneic transplantation received an infusion of a CD4/CD8 T cell-depleted leukapheresis product and Hi-Cy/Flu product as prior immunosuppressive chemotherapy, together with an infusion of an average of 2.17 × 106/kg γδ T cells with <1% CD4- or CD8-positive cells remaining in the product100. The duration of neutropenia was 20 days. Three out of the 4 patients achieved complete remission, which lasted between 2 and 8 months, while the fourth patient died from severe septicemia after day 45100. However, this patient's hematopoiesis had already recovered, and none of the patients showed any signs of acute or cGvHD nor organ injury100. Although γδ T cells have yet to be widely studied, they have shown potential for use in treatment, and subsequent research should aim to explore the safety and efficacy of γδ T cell-based immunotherapy further.
Conclusion and Future Perspectives
The importance of HSCTs in the treatment of various conditions is undeniable. However, there are several limitations of HSCTs that are worth mentioning, particularly in terms of the quantity and quality of transplanted cells and toxicity, which could lower the effectiveness of transplants in the treatment of patients. The latter could result in the need for the use of cellular engineering. In fact, several cellular engineering methods have been used to date, including UCB expansion, cell selection, infusion of immune regulatory cells, including MSCs and Tregs, and immune effector cell therapy, which includes DLIs, CAR T cells, NK cells, and γδ T cells. UCB expansion has been mostly successful, with several of the molecules used causing large increases in the number of HSPCs and improved outcomes in patients10, 12, 13, 15. In haploidentical transplantations, two cell selection methods are TCR αβ+/CD19+ cell depletion and naïve CD45RA+ cell depletion, both of which have been shown to contribute to a lower GvHD rate in patients28, 31. The use of MSCs in animal trials has led to better outcomes in mice, while the use of MSCs in patients has led to some improvements; however, treatment in humans has also lead to adverse events. Thus, further research is needed to develop safer MSC therapies associated with fewer adverse events41, 43, 48. The use of Tregs has also shown potential in lowering the rates of GvHD, but can also contribute to tumor progression while suppressing anti-tumor immunity. Thus, Tregs are not a very safe treatment option51, 52, 57. With the use of DLIs being associated with higher rates of GvHD, other immune effector cell therapies have been explored to induce GVT effects without high GvHD rates61–64. Among these, CAR T cell therapy has shown great potential in leading to higher remission and survival rates; however, several recipients have been reported to also experience adverse events71, 73. Thus, CAR T cell therapy may require some modifications to lower the rates of adverse events. Research should seek to determine the optimal way to use CAR T cells, namely whether they should be used together with or instead of HSCTs, as well as the optimal conditions for their use. While the use of NK cells is mostly safe, their use has not led to significant improvements in patient outcomes85–87. However, CAR-NK cells have shown some effectiveness in improving remission rates despite the lack of HLA matching, and the use of anti-tumor mAbs, such as rituximab, has been shown to help activate unlicensed NK cells, as well as increase their cytotoxicity88, 92. Thus, NK cells show great potential for use in these therapies. Finally, the use of γδ T cells is a more novel and less widely studied area of research that could be a potent therapy in the treatment of cancer; however, more studies are needed to confirm its safety and effectiveness99, 100.
Future research should seek to evaluate and modify cellular engineering methods to improve their safety and effectiveness in the treatment of patients. In addition, studies should also seek to explore ways in which to use genetic engineering to reduce the allogeneicity of the cells used in transplants, as well as lower the costs and improve the efficiency of the processes. As a result, universal donors and off-the-shelf therapy can be made more readily available and treatments more affordable for patients.
Ethics Approval and Consent to Participate
Not applicable
Consent for Publication
Not applicable
Availability of Data and Materials
Data sharing is not applicable to this article as no new data were generated or analyzed in this study.
Authors' Contributions
WYKH conceived the idea for the article, and KVQYS prepared the original draft of the manuscript. WYKH and KVQYS were both involved in performing the literature search and data analysis, as well as reviewing and editing the manuscript. Both authors have read and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest. Disclosure forms provided by the authors are available on the website.
References
1.Gupta V, Tallman MS, Weisdorf DJ. Allogeneic hematopoietic cell transplantation for adults with acute myeloid leukemia: Myths, controversies, and unknowns. Blood. 2011; 117: 2307-18.
2.Sullivan KM, Goldmuntz EA, Keyes-Elstein L, McSweeney PA, Pinckney A, Welch B, et al. Myeloablative autologous stem-cell transplantation for severe scleroderma. N Engl J Med. 2018; 378: 35-47.
3.Mitchell R, Nivison-Smith I, Anazodo A, Tiedemann K, Shaw PJ, Teague L, et al. Outcomes of haematopoietic stem cell transplantation for inherited metabolic disorders: a report from the Australian and New Zealand Children's Haematology Oncology Group and the Australasian Bone Marrow Transplant Recipient Registry. Pediatr Transplant. 2013; 17: 582-8.
4.Niederwieser D, Baldomero H, Atsuta Y, Aljurf M, Seber A, Greinix HT, et al. One and half million hematopoietic stem cell transplants (HSCT). Dissemination, trends and potential to improve activity by telemedicine from the Worldwide Network for Blood and Marrow Transplantation (WBMT). Blood. 2019.
5.The European Blood and Marrow Transplantation Textbook for Nurses. 2018.
6.Mehta RS, Dave H, Bollard CM, Shpall EJ. Engineering cord blood to improve engraftment after cord blood transplant. Stem Cell Investig. 2017; 4: 41.
7.Broxmeyer HE. Enhancing the efficacy of engraftment of cord blood for hematopoietic cell transplantation. Transfus Apher Sci. 2016; 54: 364-72.
8.Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013; 122: 491-8.
9.Anand S, Thomas S, Hyslop T, Adcock J, Corbet K, Gasparetto C, et al. Transplantation of ex vivo expanded umbilical cord blood (NiCord) decreases early infection and ospitalization. Biol Blood Marrow Transplant. 2017; 23: 1151-7.
10.Horwitz ME, Wease S, Blackwell B, Valcarcel D, Frassoni F, Boelens JJ, et al. Phase I/II study of stem-cell transplantation using a single cord blood unit expanded ex vivo with nicotinamide. J Clin Oncol. 2019; 37: 367-74.
11.Horwitz ME, Stiff PJ, Cutler CS, Brunstein CG, Hanna R, Maziarz RT, et al. Omidubicel versus standard myeloablative umbilical cord blood transplantation: Results of a Phase III Randomized Study. Blood. 2021; 138: 1429-40.
12.Wagner JE Jr, Brunstein CG, Boitano AE, DeFor TE, McKenna D, Sumstad D, et al. Phase I/II Trial of StemRegenin-1 expanded umbilical cord blood hematopoietic stem cells supports testing as a stand-alone graft. Cell Stem Cell. 2016; 18: 144-55.
13.Dumont-Lagacé M, Li Q, Tanguay M, Chagraoui J, Kientega T, Cardin GB, et al. UM171-expanded cord blood transplants support robust T cell reconstitution with low rates of severe infections. Transplant Cell Ther. 2021; 27: 76.e1-9.
14.Cohen S, Roy J, Lachance S, Delisle JS, Marinier A, Busque L, et al. Hematopoietic stem cell transplantation using single UM171-expanded cord blood: a single-arm, phase 1-2 safety and feasibility study. Lancet Haematol. 2020; 7: e134-45.
15.Bari S, Zhong Q, Fan X, Poon Z, Lim AST, Lim TH, et al. Ex vivo expansion of CD34+CD90+CD49f+ hematopoietic stem and progenitor cells from non-enriched umbilical cord blood with azole compounds. Stem Cells Transl Med. 2018; 7: 376-93.
16.Mehta RS, Saliba RM, Cao K, Kaur I, Rezvani K, Chen J, et al. Ex vivo mesenchymal precursor cell-expanded cord blood transplantation after reduced-intensity conditioning regimens improves time to neutrophil recovery. Biol Blood Marrow Transplant. 2017; 23: 1359-66.
17.Ratajczak MZ, Suszynska M. Emerging strategies to enhance homing and engraftment of hematopoietic stem cells. Stem Cell Rev Rep. 2016; 12: 121-8.
18.Kumar S, Geiger H. HSC niche biology and HSC expansion ex vivo. Trends Mol Med. 2017; 23: 799-819.
19.Constantinou VC, Bouinta A, Karponi G, Zervou F, Papayanni PG, Stamatoyannopoulos G, et al. Poor stem cell harvest may not always be related to poor mobilization: lessons gained from a mobilization study in patients with β-thalassemia major. Transfusion. 2017; 57: 1031-9.
20.Seshadri T, Al-Farsi K, Stakiw J, Ma C, Saragosa R, Franke N, et al. G-CSF-stimulated BM progenitor cells supplement suboptimal peripheral blood hematopoietic progenitor cell collections for auto transplantation. Bone Marrow Transplant. 2008; 42: 733-7.
21.Aggarwal R, Lu J, Pompili VJ, Das H. Hematopoietic stem cells: transcriptional regulation, ex vivo expansion and clinical application. Curr Mol Med. 2012; 12: 34-49.
22.Tajer P, Pike-Overzet K, Arias S, Havenga M, Staal FJT. Ex vivo expansion of hematopoietic stem cells for therapeutic purposes: lessons from development and the niche. Cells. 2019; 8: 169.
23.Baron F, Nagler A. Novel strategies for improving hematopoietic reconstruction after allogeneic hematopoietic stem cell transplantation or intensive chemotherapy. Expert Opin Biol Ther. 2017; 17: 163-74.
24.Farhadfar N, Hogan WJ. Overview of the progress on haploidentical hematopoietic transplantation. World J Transplant. 2016; 6: 665-74.
25.Sahasrabudhe K, Otto M, Hematti P, Kenkre V. TCR αβ+/CD19+ cell depletion in haploidentical hematopoietic allogeneic stem cell transplantation: a review of current data. Leuk Lymphoma. 2019; 60: 598-609.
26.Mitchell R, Cole T, Shaw PJ, Mechinaud F, O'Brien T, Fraser C. TCR α+β+/CD19+ cell-depleted hematopoietic stem cell transplantation for pediatric patients. Pediatr Transplant. 2019; 23: e13517.
27.Airoldi I, Bertaina A, Prigione I, Zorzoli A, Pagliara D, Cocco C, et al. γδ T-cell reconstitution after HLA-haploidentical hematopoietic transplantation depleted of TCR-αβ+/CD19+ lymphocytes. Blood. 2015; 125: 2349-58.
28.Balashov D, Shcherbina A, Maschan M, Trakhtman P, Skvortsova Y, Shelikhova L, et al. Single-Center Experience of Unrelated and Haploidentical Stem Cell Transplantation with TCRαβ and CD19 Depletion in Children with Primary Immunodeficiency Syndromes. Biol Blood Marrow Transplant. 2015; 21: 1955-62.
29.Bleakley M, Gooley TA, Hilzinger B, Riddell SR, Shlomchik WD. NaïVe T Cell depletion of PBSC grafts results in very low rates of chronic Gvhd and high survival. Blood. 2016; 128: 668.
30.Diaz MA, Molina B, Sebastian E, Galvez E, Zubicaray J, Sevilla J, et al. CD45 RA depletion as an allogeneic hematopoietic transplantation platform in children from HLA-identical donors. Biol Blood Marrow Transplant. 2019; 25: S205-6.
31.Triplett BM, Muller B, Kang G, Li Y, Cross SJ, Moen J, et al. Selective T-cell depletion targeting CD45RA reduces viremia and enhances early T-cell recovery compared with CD3-targeted T-cell depletion. Transpl Infect Dis. 2018; 20: e12823.
32.Touzot F, Neven B, Dal-Cortivo L, Gabrion A, Moshous D, Cros G, et al. CD45RA depletion in HLA-mismatched allogeneic hematopoietic stem cell transplantation for primary combined immunodeficiency: A preliminary study. J Allergy Clin Immunol. 2015; 135: 1303-9.e1-3.
33.Shook DR, Triplett BM, Eldridge PW, Kang G, Srinivasan A, Leung W. Haploidentical stem cell transplantation augmented by CD45RA negative lymphocytes provides rapid engraftment and excellent tolerability. Pediatr Blood Cancer. 2015; 62: 666-73.
34.Bleakley M, Heimfeld S, Loeb KR, Jones LA, Chaney C, Seropian S, et al. Outcomes of acute leukemia patients transplanted with naive T cell-depleted stem cell grafts. J Clin Invest. 2015; 125: 2677-89.
35.Rådestad E, Sundin M, Törlén J, Thunberg S, Önfelt B, Ljungman P, et al. Individualization of hematopoietic stem cell transplantation using alpha/beta T-cell depletion. Front Immunol. 2019; 10: 189.
36.Shasheleva DA, Maschan AA, Shelikhova LN, Petrova UN, Kurnikova EE, Illarionova OI, et al. Hematopoietic stem cell transplantation with alpha/beta T-lymphocyte depletion and short course of eculizumab in adolescents and young adults with paroxysmal nocturnal hemoglobinuria. Ter Arkh. 2018; 90: 57-64.
37.Horwitz EM, Andreef M, Frassoni F. Mesenchymal stromal cells. Biol Blood Marrow Transplant. 2007; 13: 53-7.
38.Wu X, Jiang J, Gu Z, Zhang J, Chen Y, Liu X. Mesenchymal stromal cell therapies: Immunomodulatory properties and clinical progress. Stem Cell Res Ther. 2020; 11: 345.
39.Fitzsimmons REB, Mazurek MS, Soos A, Simmons CA. Mesenchymal stromal/stem cells in regenerative medicine and tissue engineering. Stem Cells Int. 2018; 2018: 8031718.
40.Burnham AJ, Daley-Bauer LP, Horwitz EM. Mesenchymal stromal cells in hematopoietic cell transplantation. Blood Adv. 2020; 4: 5877-87.
41.Gonzaga VF, Wenceslau CV, Lisboa GS, Frare EO, Kerkis I. Mesenchymal stem cell benefits observed in bone marrow failure and acquired aplastic anemia. Stem Cells Int. 2017; 2017: 8076529.
42.Bueno C, Roldan M, Anguita E, Romero-Moya D, Martín-Antonio B, Rosu-Myles M, et al. Bone marrow mesenchymal stem cells from patients with aplastic anemia maintain functional and immune properties and do not contribute to the pathogenesis of the disease. Haematologica. 2014; 99: 1168-75.
43.Cetin M, Akyol G, Gonen ZB, Keklik M, Zararsiz G, Unal A, et al. Additional infusions of mesenchymal stem cells improve response rate in multidrug-resistant GvHD patients. Bone Marrow Transplant. 2017; 52: 783-5.
44.Salmenniemi U, Itälä-Remes M, Nystedt J, Putkonen M, Niittyvuopio R, Vettenranta K, et al. Good responses but high TRM in adult patients after MSC therapy for GvHD. Bone Marrow Transplant. 2017; 52: 606-8.
45.Zhao L, Chen S, Yang P, Cao H, Li L. The role of mesenchymal stem cells in hematopoietic stem cell transplantation: Prevention and treatment of graft-versus-host disease. Stem Cell Res Ther. 2019; 10: 182.
46.Dotoli GM, De Santis GC, Orellana MD, de Lima Prata K, Caruso SR, Fernandes TR, et al. Mesenchymal stromal cell infusion to treat steroid-refractory acute GvHD III/IV after hematopoietic stem cell transplantation. Bone Marrow Transplant. 2017; 52: 859-62.
47.Liu Z, Zhang Y, Xiao H, Yao Z, Zhang H, Liu Q, et al. Cotransplantation of bone marrow-derived mesenchymal stem cells in haploidentical hematopoietic stem cell transplantation in patients with severe aplastic anemia: An interim summary for a multicenter phase II trial results. Bone Marrow Transplant. 2017; 52: 704-10.
48.Fan X, Guo D, Cheung AMS, Poon ZY, Yap CS, Goh SE, et al. Mesenchymal Stromal Cell (MSC)-Derived Combination of CXCL5 and Anti-CCL24 Is Synergistic and Superior to MSC and Cyclosporine for the Treatment of Graft-versus-Host Disease. Biol Blood Marrow Transplant. 2018; 24: 1971-80.
49.Wang X, Zhang M, He P. Pre-infusion single-dose mesenchymal stem cells promote platelet engraftment and decrease severe acute graft versus host disease without relapse in haploidentical peripheral blood stem cell transplantation. J Int Med Res. 2020; 48: 300060520920438.
50.Pang Y, Xiao HW, Zhang H, Liu ZH, Li L, Gao Y, et al. Allogeneic Bone Marrow-Derived Mesenchymal Stromal Cells Expanded In Vitro for Treatment of Aplastic Anemia: A Multicenter Phase II Trial. Stem Cells Transl Med. 2017; 6: 1569-75.
51.Zaini RG, Al-Rehaili AA. The Therapeutic Strategies of Regulatory T cells in malignancies and stem cell transplantations. J Oncol. 2019; 2019: 5981054.
52.Yang S, Sheng X, Xiang D, Wei X, Chen T, Yang Z, et al. CD150highTreg cells may attenuate graft versus host disease and intestinal cell apoptosis after hematopoietic stem cell transplantation. Am J Transl Res. 2019; 11: 1299-310. eCollection 2019.
53.Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci. 2019; 110: 2080-9.
54.Yang J, Fan H, Hao J, Ren Y, Chen L, Li G, et al. Amelioration of acute graft-versus-host disease by adoptive transfer of ex vivo expanded human cord blood CD4+CD25+ forkhead box protein 3+ regulatory T cells is associated with the polarization of Treg/Th17 balance in a mouse model. Transfusion. 2012; 52: 1333-47.
55.Lim JY, Park MJ, Im KI, Kim N, Jeon EJ, Kim EJ, et al. Combination cell therapy using mesenchymal stem cells and regulatory T-cells provides a synergistic immunomodulatory effect associated with reciprocal regulation of Th1/Th2 and Th17/Treg cells in a murine acute graft-versus-host disease model. Cell Transplant. 2014; 23: 703-14.
56.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017; 27: 109-18.
57.Brunstein CG, Miller JS, McKenna DH, Hippen KL, DeFor TE, Sumstad D, et al. Umbilical cord blood-derived T regulatory cells to prevent GVHD: Kinetics, toxicity profile, and clinical effect. Blood. 2016; 127: 1044-51.
58.Shaffer BC, Hsu KC. How important is NK alloreactivity and KIR in allogeneic transplantation? Best Pract Res Clin Haematol. 2016; 29: 351-8.
59.Liou A, Wahlstrom JT, Dvorak CC, Horn BN. Safety of pre-emptive donor lymphocyte infusions (DLI) based on mixed chimerism (MC) in peripheral blood or bone marrow subsets in children undergoing hematopoietic stem cell transplant (HSCT) for hematologic malignancies. Bone Marrow Transplant. 2017; 52: 1057-9.
60.Swaminathan VV, Uppuluri R, Patel S, Sivashankaran M, Ravichandran N, Ramanan KM, et al. Safety and efficacy of fresh whole blood donor lymphocyte infusion in children. Bone Marrow Transplant. 2019; 54: 1892-7.
61.Davies JK, Brennan LL, Wingard JR, Cogle CR, Kapoor N, Shah AJ, et al. Infusion of alloanergized donor lymphocytes after CD34-selected haploidentical myeloablative hematopoietic stem cell transplantation. Clin Cancer Res. 2018; 24: 4098-109.
62.Marks DI, Lush R, Cavenagh J, Milligan DW, Schey S, Parker A, et al. The toxicity and efficacy of donor lymphocyte infusions given after reduced-intensity conditioning allogeneic stem cell transplantation. Blood. 2002; 100: 3108-14.
63.Yan CH, Liu QF, Wu DP, Zhang X, Xu LP, Zhang XH, et al. Prophylactic donor lymphocyte infusion (DLI) followed by minimal residual disease and graft-versus-host disease-guided multiple DLIs could improve outcomes after allogeneic hematopoietic stem cell transplantation in patients with refractory/relapsed acute. Biol Blood Marrow Transplant. 2017; 23: 1311-9.
64.Nikiforow S, Kim HT, Daley H, Reynolds C, Jones KT, Armand P, et al. A phase I study of CD25/regulatory T-cell-depleted donor lymphocyte infusion for relapse after allogeneic stem cell transplantation. Haematologica. 2016; 101: 1251-9.
65.Gao XN, Lin J, Wang SH, Huang WR, Li F, Li HH, et al. Donor lymphocyte infusion for prevention of relapse after unmanipulated haploidentical PBSCT for very high-risk hematologic malignancies. Ann Hematol. 2019; 98: 185-93.
66.Kerbage F, Sakr R, Lapierre V, Alexandrova K, Coman T, Leroux S, et al. Donor lymphocyte infusions after allogeneic transplantation: A Single-Center Experience. Clin Lymphoma Myeloma Leuk. 2020; 20: 209-11.
67.Merker M, Salzmann-Manrique E, Katzki V, Huenecke S, Bremm M, Bakhtiar S, et al. Clearance of hematologic malignancies by allogeneic cytokine-induced killer cell or donor lymphocyte Infusions. Biol Blood Marrow Transplant. 2019; 25: 1281-92.
68.Maude SL, Teachey DT, Porter DL, Grupp SA. CD19-targeted chimeric antigen receptor T-cell therapy for acute lymphoblastic leukemia. Blood. 2015; 125: 4017-23.
69.Hay KA, Turtle CJ. Chimeric antigen receptor (CAR) T cells: lessons learned from targeting of CD19 in B-cell malignancies. Drugs. 2017; 77: 237-45.
70.Brudno JN, Kochenderfer JN. Toxicities of chimeric antigen receptor T cells: recognition and management. Blood. 2016; 127: 3321-30.
71.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H, et al. Tisagenlecleucel in children and young adults with B-Cell lymphoblastic leukemia. N Engl J Med. 2018; 378: 439-48.
72.Turtle CJ, Hanafi LA, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin's lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016; 8: 355ra116.
73.Wang X, Popplewell LL, Wagner JR, Naranjo A, Blanchard MS, Mott MR, et al. Phase 1 studies of central memory-derived CD19 CAR T-cell therapy following autologous HSCT in patients with B-cell NHL. Blood. 2016; 127: 2980-90.
74.Jiang H, Li C, Yin P, Guo T, Liu L, Xia L, et al. Anti-CD19 chimeric antigen receptor-modified T-cell therapy bridging to allogeneic hematopoietic stem cell transplantation for relapsed/refractory B-cell acute lymphoblastic leukemia: an open-label pragmatic clinical trial. Am J Hematol. 2019; 94: 1113-22.
75.Schuster SJ, Svoboda J, Chong EA, Nasta SD, Mato AR, Anak Ö, et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N Engl J Med. 2017; 377: 2545-54.
76.Enblad G, Karlsson H, Gammelgård G, Wenthe J, Lövgren T, Amini RM, et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin Cancer Res. 2018; 24: 6185-94.
77.Curran KJ, Margossian SP, Kernan NA, Silverman LB, Williams DA, Shukla N, et al. Toxicity and response after CD19-specific CAR T-cell therapy in pediatric/young adult relapsed/refractory B-ALL. Blood. 2019; 134: 2361-8.
78.Sauter CS, Senechal B, Rivière I, Ni A, Bernal Y, Wang X, et al. CD19 CAR T cells following autologous transplantation in poor-risk relapsed and refractory B-cell non-Hodgkin lymphoma. Blood. 2019; 134: 626-35.
79.Ma F, Ho JY, Du H, Xuan F, Wu X, Wang Q, et al. Evidence of long-lasting anti-CD19 activity of engrafted CD19 chimeric antigen receptor-modified T cells in a phase I study targeting pediatrics with acute lymphoblastic leukemia. Hematol Oncol. 2019; 37: 601-8.
80.Park JH, Rivière I, Gonen M, Wang X, Sénéchal B, Curran KJ, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. N Engl J Med. 2018; 378: 449-59.
81.Cooley S, Parham P, Miller JS. Strategies to activate NK cells to prevent relapse and induce remission following hematopoietic stem cell transplantation. Blood. 2018; 131: 1053-62.
82.Hattori N, Nakamaki T. Natural killer immunotherapy for minimal residual disease eradication following allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia. Int J Mol Sci. 2019; 20: 2057.
83.Lee DA, Denman CJ, Rondon G, Woodworth G, Chen J, Fisher T, et al. Haploidentical natural killer cells infused before allogeneic stem cell transplantation for myeloid malignancies: A Phase I Trial. Biol Blood Marrow Transplant. 2016; 22: 1290-8.
84.Shah N, Li L, McCarty J, Kaur I, Yvon E, Shaim H, et al. Phase I study of cord blood-derived natural killer cells combined with autologous stem cell transplantation in multiple myeloma. Br J Haematol. 2017; 177: 457-66.
85.Nguyen R, Wu H, Pounds S, Inaba H, Ribeiro RC, Cullins D, et al. A phase II clinical trial of adoptive transfer of haploidentical natural killer cells for consolidation therapy of pediatric acute myeloid leukemia. J Immunother Cancer. 2019; 7: 81.
86.Choi I, Yoon SR, Park SY, Kim H, Jung SJ, Jang YJ, et al. Donor-derived natural killer cells infused after human leukocyte antigen-haploidentical hematopoietic cell transplantation: A Dose-Escalation Study. Biol Blood Marrow Transplant. 2014; 20: 696-704.
87.Ciurea SO, Schafer JR, Bassett R, Denman CJ, Cao K, Willis D, et al. Phase 1 clinical trial using mbIL21 ex vivo-expanded donor-derived NK cells after haploidentical transplantation. Blood. 2017; 130: 1857-68.
88.Liu E, Marin D, Banerjee P, Macapinlac HA, Thompson P, Basar R, et al. Use of CAR-Transduced Natural Killer Cells in CD19-Positive Lymphoid Tumors. N Engl J Med. 2020; 382: 545-53.
89.Liu E, Tong Y, Dotti G, Shaim H, Savoldo B, Mukherjee M, et al. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018; 32: 520-31.
90.Wang W, Erbe AK, Hank JA, Morris ZS, Sondel PM. NK cell-mediated antibody-dependent cellular cytotoxicity in cancer immunotherapy. Front Immunol. 2015; 6: 368.
91.Dixon KJ, Wu J, Walcheck B. Engineering anti-tumor monoclonal antibodies and fc receptors to enhance ADCC by human NK cells. Cancers (Basel). 2021; 13: 312.
92.Du J, Lopez-Verges S, Pitcher BN, Johnson J, Jung SH, Zhou L, et al. CALGB 150905 (Alliance): rituximab broadens the antilymphoma response by activating unlicensed NK cells. Cancer Immunol Res. 2014; 2: 878-89.
93.Shaffer BC, Le Luduec JB, Forlenza C, Jakubowski AA, Perales MA, Young JW, et al. Phase II study of haploidentical natural killer cell infusion for treatment of relapsed or persistent myeloid malignancies following allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2016; 22: 705-9.
94.Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016; 8: 357ra123.
95.Bachanova V, Sarhan D, DeFor TE, Cooley S, Panoskaltsis-Mortari A, Blazar BR, et al. Haploidentical natural killer cells induce remissions in non-Hodgkin lymphoma patients with low levels of immune-suppressor cells. Cancer Immunol Immunother. 2018; 67: 483-94.
96.Zou C, Zhao P, Xiao Z, Han X, Fu F, Fu L. γδ T cells in cancer immunotherapy. Oncotarget. 2017; 8: 8900-9.
97.Zhao Y, Niu C, Cui J. Gamma-delta (γδ) T Cells: friend or foe in cancer development. J Transl Med. 2018; 16: 3.
98.Raverdeau M, Cunningham SP, Harmon C, Lynch L. γδ T cells in cancer: a small population of lymphocytes with big implications. Clin Transl Immunology. 2019; 8: e01080.
99.Zhou J, Kang N, Cui L, Ba D, He W. Anti-γδ TCR antibody-expanded γδ T cells: a better choice for the adoptive immunotherapy of lymphoid malignancies. Cell Mol Immunol. 2012; 9: 34-44.
100.Wilhelm M, Smetak M, Schaefer-Eckart K, Kimmel B, Birkmann J, Einsele H, et al. Successful adoptive transfer and in vivo expansion of haploidentical γδ T cells. J Transl Med. 2014; 12: 45.
Search
News


