Volume 4 (2021) Issue 2 No.2 Pages 29-34
Abstract
We report herein haplo-identical hematopoietic stem cell transplantation (haplo-HSCT) by T-cell replete graft infusion, with post-transplant cyclophosphamide (PTCy) in patients with hemoglobinopathies. Patients received a conditioning regimen consisting of either busulfan, fludarabine, cyclophosphamide, with antithymocyte globulin or Thiotepa, antithymocyte globulin, fludarabine, cyclophosphamide, and TBI. The median follow-up period was 14.3 months (range, 1-63 months). Overall survival (OS) and disease-free survival (DFS) were 80% and 62.8%, respectively. Incidence of secondary graft failure was 14%. Incidences of acute graft-versus-host disease (aGvHD) and chronic graft-versus-host disease (cGvHD) were 22.5% and 20%, respectively. Cytomegalovirus (CMV) reactivation was observed in 42.5% of cases. The 100-day mortality rate was 20%, with sepsis and aGvHD being the predominant causes of death.
Introduction
Hematopoietic stem cell transplantation (HSCT) from an HLA-matched related or unrelated donor is the only curative option for hemoglobinopathies such as thalassemia major (TM) and sickle cell disease (SCD) 1,2. However, only 25% of patients have an HLA-matched sibling donor. The chance of finding a fully matched unrelated donor in the western population according to a recent study from the US National Marrow Donor Program was around 50%3. In developing countries, this option is less feasible due to the high cost of procurement, and the limited pools of donors among particular ethnic groups. Since every patient shares exactly one HLA haplotype with each biological parent or child, and one half with siblings, an eligible haplo donor can be rapidly identified in nearly all cases4.
Historically, graft rejection and high rates of graft versus host disease (GvHD) have been the major obstacles to haplo-HSCT. The development of strategies that result in selective T cell depletion, such as graft engineering using TCR alpha/beta+, CD19+ deplete grafts, or in vivo depletion using post-transplant high-dose cyclophosphamide (PTCy) have significantly improved outcomes5,6. Nevertheless, graft failure, high treatment-related mortality (TRM) due to infections or GvHD, and delayed immune reconstitution remain significant concerns7. With increasing experience with haplo-HSCT, some centers are reporting clinical outcomes comparable to transplants with fully matched unrelated donors8,9. We report here our experience with Haplo-HSCT in patients with hemoglobinopathies.
Patients and Methods
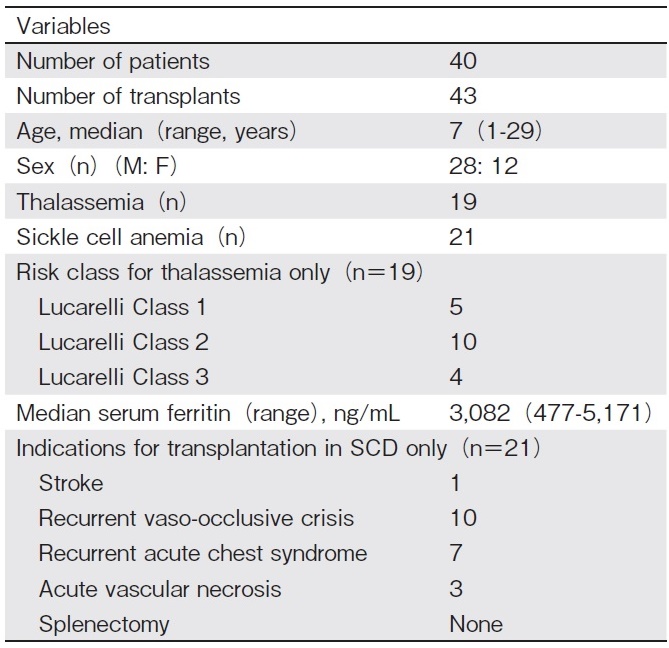
This is a retrospective, single-center study that evaluates the outcomes of haplo-HSCT in 40 consecutive patients with hemoglobinopathies (TM and SCD) who underwent 43 T-cell replete transplants between September 2014 and December 2019 at BLK Superspeciality Hospital, New Delhi. Patients were eligible for haplo- HSCT if they did not have any HLA-matched related donor, or no unrelated donor. Indications for transplant for SCD were recurrent vaso-occlusive crises, acute vas- cular necrosis of the hip joint, acute chest crises, stroke, and transfusion dependence in TM. Out of a total of 40 patients with hemoglobinopathies, 19 received transplants for TM, and 21 for SCD. Three TM patients underwent a second transplant due to primary graft failure. Data were collected from the hospital database regarding patient and transplant characteristics (Table 1 and 2). Pre-transplant evaluation of all patients and donors was performed as per established protocol. Before transplantation, most TM patients were heavily transfused and had moderate to severe iron overload, as evidenced by high median serum ferritin levels. This study was approved by the Institutional Review Board and Ethics Committee of the hospital, as per the Declaration of Helsinki.

All TM patients received 2 courses of fludarabine (40 mg/m2 ? for 5 days) followed by dexamethasone (25 mg/m2 ? for 5 days) one month apart on day-68 to day-64 and day-40 to day-36 to achieve pre-transplant immunosuppression (PTIS) to suppress host T cell function and facilitate engraftment, as has been advocated by Anurathapan et al10. All transplant recipients were screened for donor-specific antibodies (DSA) by single bead assay on Luminex (Lifecodes LSA single antigen class ? & Class ? kit; Immucor, USA) before transplant. If antibody levels were high (median fluorescence intensity[MFI]>2,000), patients were desensitized using rituximab (375 mg/m2) or plasmapheresis, as per desensitization protocol. DSA was re-checked prior to HSCT.
Patients were admitted to high-efficiency particulate air (HEPA) filter positive-pressure isolation rooms for HSCT. Oral valacyclovir was administered as anti-viral prophylaxis for herpes simplex virus, and oral fluconazole as antifungal prophylaxis from day+1. Ursodeoxycholic acid (5 mg/kg/dose every 8 hours) was administered from the onset of conditioning for regimen related hepatic toxicity/sinusoidal obstruction syndrome (SOS) prophylaxis11. Granulocyte colony stimulating factor (G-CSF) was started from day+5 (5μg/kg/dose once daily) SC or ? until absolute neutrophil count (ANC) was>1.0×106/μL. Neutropenic sepsis was treated with ? antibiotics as per institutional policy. SOS was diagnosed and classified as mild, moderate, or severe according to the New EBMT criteria12. Acute GvHD was graded according to the modified Seattle Glucksberg criteria13.
Human leukocyte antigen (HLA) typing was performed with a high-resolution PCR based method (HLA A, B, C, DRB1, and DQB1). Donors were selected based on DSA status, CMV sero status, age, sex, blood group, and degree of haplo match. Donor peripheral blood stem cells (PBSC) were mobilized with granulocyte colony-stimulating factor (10μg/kg/day) from day-4 until collection on day-1 and day 0 (if stem cell collection was inadequate). Bone marrow harvest was performed under general anesthesia on day 0.
The initial six patients with SCD received BM as a stem cell source. One patient received a top up with PBSC, as the BM contained an inadequate concentration of cells. All patients received un-manipulated stem cells on day 0. Red blood cells and plasma were depleted in bone marrow products in cases of ABO mismatch.
For all patients with TM, the conditioning regimen consisted of ? busulfan/fludarabine/anti thymocyte globulin (3.2 mg/kg/day intravenous busulfan from day-7 to day-4; 35 mg/m2/day intravenous fludarabine from day-7 to day-2, and 1.5 mg/kg/day intravenous thymoglobulin from day-11 to day-9). The first 10 patients with SCD undergoing Haplo HSCT received Thiotepa/Thymoglobulin/Fludarabine/Cyclophosphamide/ TBI (one 5 mg/kg dose of intravenous Thiotepa on day-7, 30 mg/m2 intravenous fludarabine from day-6 to day-2, 14.5 mg/kg intravenous cyclophosphamide on day-5 to day-4, and 200 cGy TBI on day-1). The protocol was changed to Fludarabine/Busulfan/Thymoglobulin for the remaining 11 patients with SCD, as significant primary graft rejection was observed. In 3 patients who underwent a second transplant due to graft failure, the regimen was fludarabine/cyclophosphamide/ TBI (30 mg/m2/dose intravenous fludarabine for 5 days, 14.5 mg/kg intravenous cyclophosphamide for 2 days, and 2,400 cGy doses of TBI).
All patients received PTCy with 50 mg/kg/day for GvHD prophylaxis on days+3 and+4, with oral tacrolimus ( 0.06 mg/kg twice daily) and oral mycophenolate sodium (10 mg/kg/dose three times daily) from day+5. Hyper-hydration together with mesna was administered with cyclophosphamide. Serum tacrolimus trough levels were monitored regularly and maintained at 5-15 ng/mL for 6-9 months post-transplant. Mycophenolate sodium was continued until day+35, then tapered off by day+60.
Chimerism analysis was performed regularly on peripheral blood samples from Day+21 onwards. Donor-recipient DNA percentages were determined by PCR-based analysis of short tandem repeats using a CD3500DX Genetic analyzer.
SPSS 25 (IBM Corp., USA) was used to perform statistical analyses. Survival analyses were performed using the Kaplan-Meier test. Univariate analyses of risk factors for HSCT were performed using the Cox regression test, and p-values<0.05 were considered to indicate statistical significance.
Results
The median CD34+stem cell dose was 10.7×106cells/ kg (range, 1.67-20.4 cells/kg). The median times to neutrophil and platelet engraftment were 14 days (range, 11-24 days) and 17 days (range 11-35 days), respectively (Table 3). The median follow-up was 14.3 months (range, 1-63 months). Of the 40 patients, who underwent 43 transplants, 37 underwent primary engraftment, 2 expired before engraftment, and 4 (9%) experienced primary graft failure (TM-3, SCD-1). Six (14%) patients (0ne TM, SCD-5) experienced secondary graft failure after a median of 55 days (range, 34-64 days). Of these six, one patient died due to sepsis. The others remained alive with primary disease. Of the 4 patients who experienced primary graft failure, three underwent a second transplant. Two of these three became well, and the third expired. The fourth patient experienced autologous hematopoietic recovery and remained alive with the primary disease. Autologous hematopoiesis was achieved in all 6 patients who experienced secondary graft failure.
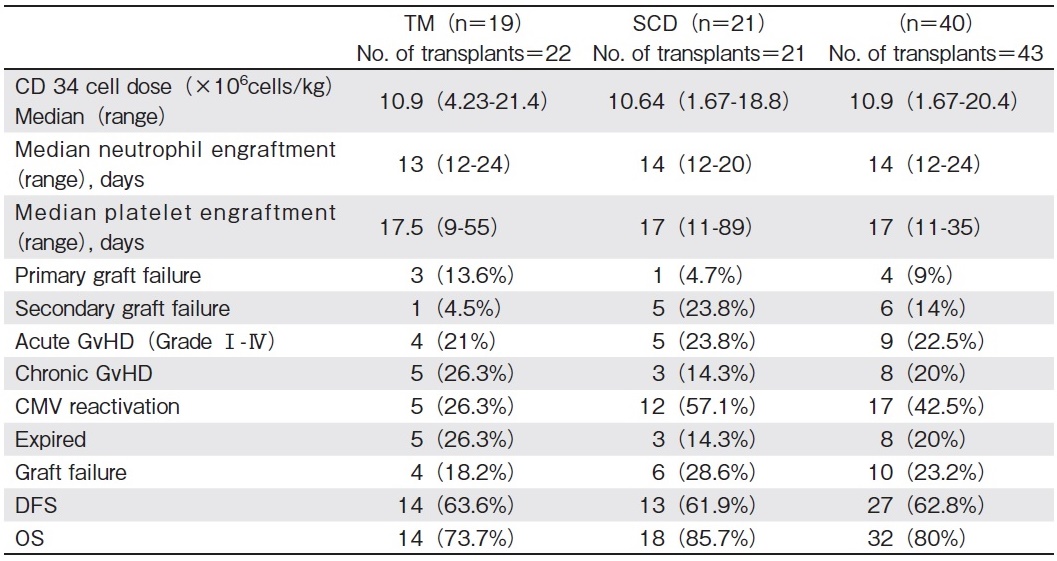
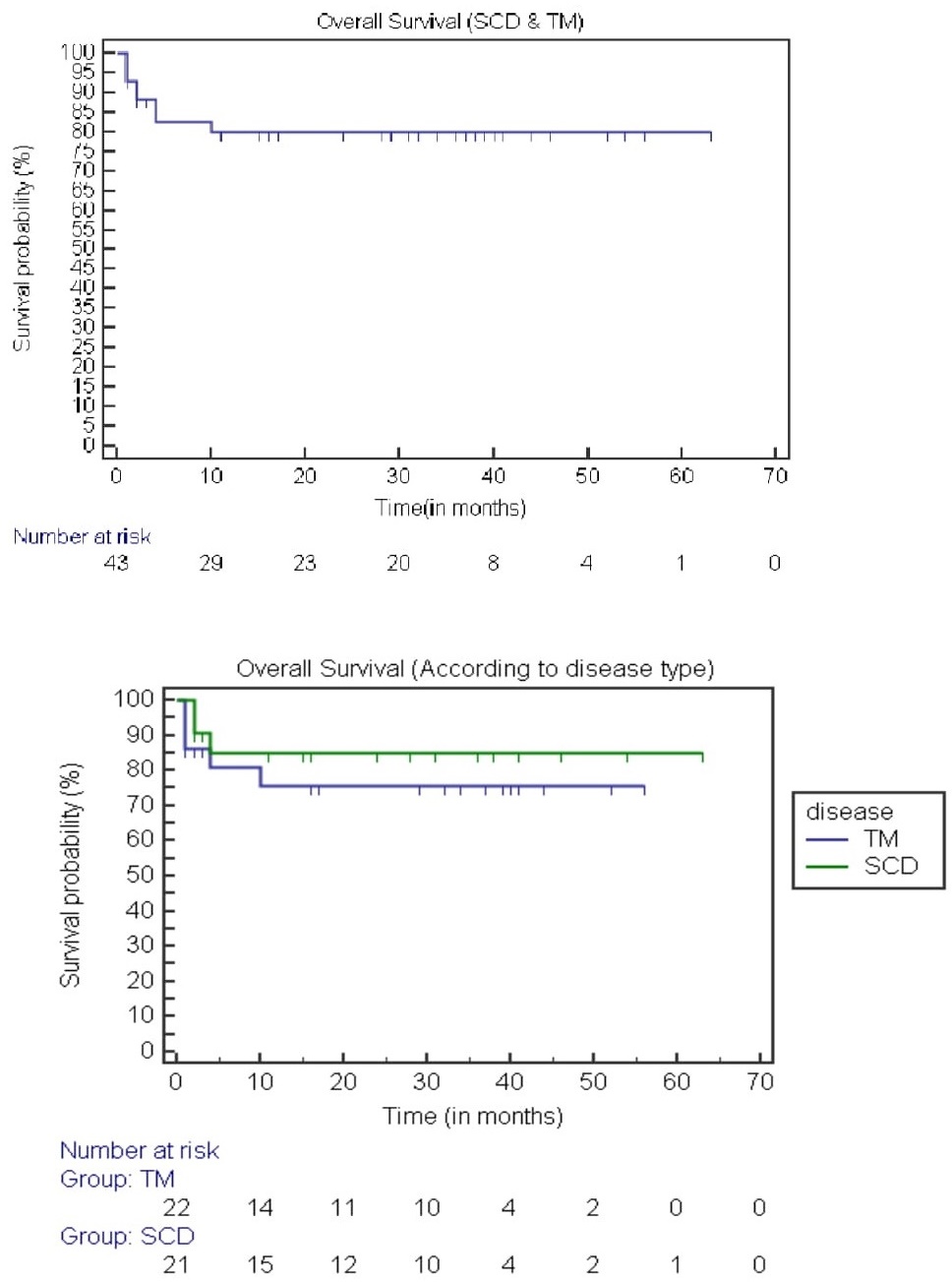
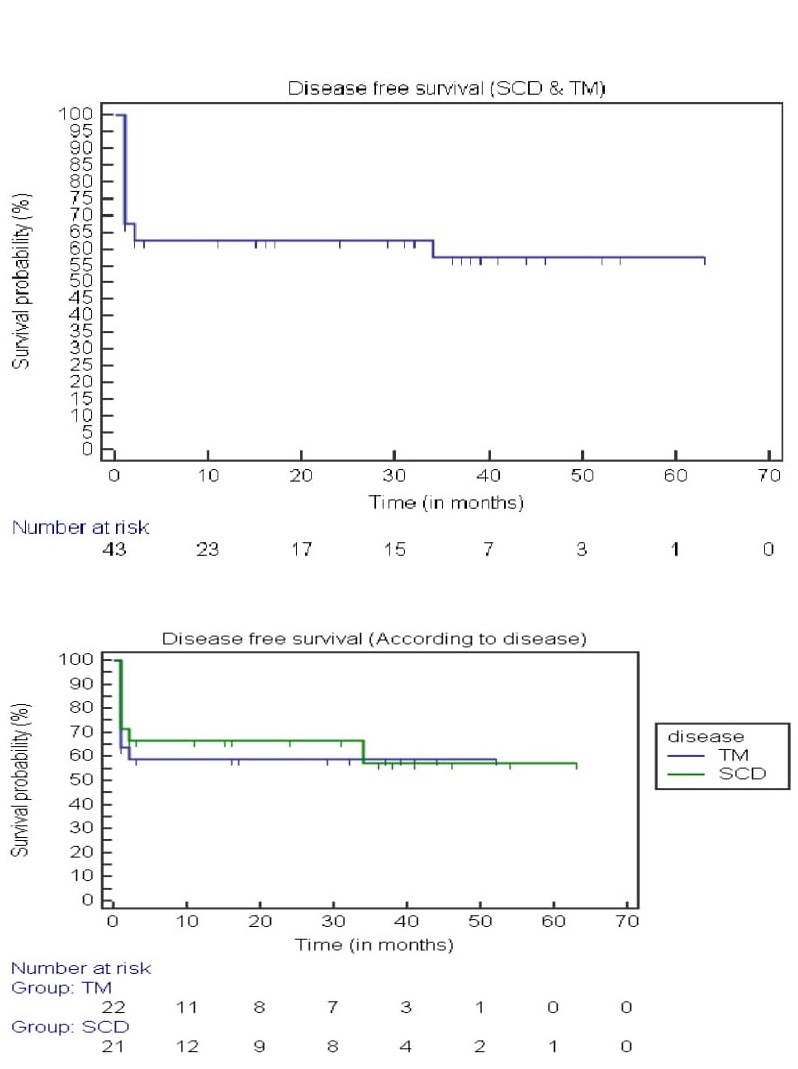
Twenty-one (49%) patients developed grade 2-3 mucositis. Three patients (7%) developed mild to moderate reversible SOS of the liver, and 5 (12.5%) developed hemorrhagic cystitis. Posterior reversible encephalopathy syndrome (PRES) was observed in 7 patients (17.5%). Six of these patients had SCD (29%), which resolved completely without neurological sequelae. Five patients developed aGvHD grade ?-?, and 4 patients developed aGvHD grade ?-? (total aGvHD 22.5%). There were 2 GvHD-related deaths. Eight patients (20%) developed cGvHD. Six of these experienced limited cGvHD of the oral cavity, while 2 developed extensive cGvHD (both had a history of aGvHD). Seventeen patients (42.5%) experienced cytomegalovirus (CMV) reactivation and were treated with Gancyclovir. None of these patients developed CMV disease. No patients developed posttransplant lymphoproliferative disorder (PTLD). Twentyseven ( 63%) of our patients displayed complete donor chimerism (range 95%-99%). No patients displayed mixed chimerism. Patients who presented with mixed chimerism subsequently experienced secondary graft failure. There were a total of 6 (15%) sepsis-related deaths. Two patients died due to sepsis before engraftment, and four died due to sepsis after engraftment. Of the 5 patients with TM who expired, 3 were Class ? (high risk) and 2 were>15 years of age. Overall survival (OS) and disease-free survival (DFS) were 80% and 62.8%, respectively, at a median of 14.3 months (Figure 1 and 2). Treatment-related mortality (TRM) was 20%.
Discussion
Allogeneic HSCT remains the only potentially curative therapy for severe hemoglobinopathies like TM and SCD14,15. Expanding the donor pool through the use of related HLA-haploidentical donors, most patients in need of HSCT become eligible.
An initial study of Haplo-HSCT in TM using busulfan, cyclophosphamide, and/or antilymphocyte globulin and/ or TBI, and administering cyclosporine and methotrexate with or without methyl prednisolone for GvHD prophylaxis showed disappointing results, with a high rate of graft failure (55%), severe aGVHD and cGvHD (37% and 47%) and TRM (34%) 16. Subsequent use of nonmyeloablative conditioning together with PTCy in haplo- HSCT for malignant conditions yielded 13% graft failure, 34% aGvHD ?-?, and respective OS and EFS rates of 36% and 26%5. A similar non myeloablative regimen was assessed later for SCD that achieved a reduction in aGvHD, but displayed a graft failure rate of approximately 40-50%17. Increasing the TBI dose to 400 Gy and following the same protocol for TM reduced the primary graft failure rate to 6%, with 18% of patients developing unstable mixed chimerism, necessitating the use of longterm immunosuppressive therapy. No mortality was reported in this study, and aGvHD and cGvHD rates were 29% and 18%, respectively18.
In a study of SCD, 15 patients received haplo HSCT following the John Hopkins non-myeloablative haplo protocol with Thiotepa. Ninety three percent of patients had>95% stable donor engraftment at 6 months, with 100% OS. Median times to neutrophil and platelet engraftment were 22 days and 28 days, respectively. Two patients had grade ?-? acute GVHD, and 1 patient had mild chronic GVHD19. We used the same protocol in initial treatment of 10 patients with SCD, and observed a graft failure rate of 40%.
Some groups have used T cell depletion to reduce GvHD in haplo-HSCT. In a study including 22 TM patients, graft rejection and TRM were 23% and 7%, respectively6. Gaziev et al. reported 14% graft failure, with 84% and 69% OS and DFS, respectively. Rates of aGvHD (?-?) and cGvHD were 28% and 21%, respectively. However, the PTLD rate was significantly elevated (23%), suggesting that although this study demonstrated a low graft failure rate, there was morbidity and mortality associated with delayed immune reconstitution20.
Anurathapan et al. have reported outcomes for 31 TM patients who underwent T cell-replete peripheral blood haploidentical HSCT, receiving 2 cycles of PTIS therapy, with PTCy as GvHD prophylaxis10. The primary graft failure rate was 6.5%. Twenty nine percent developed aGVHD grade ?-?, and 16% developed limited cGVHD. Two-year OS and EFS were 95% and 94%, respectively. In our TM cohort, comprising 19 patients, the primary graft failure rate was 13.5%. Twenty one percent developed aGVHD grade ?-?, and 26% developed limited cGVHD10. OS and EFS were 64% and 74%, respectively. The reason for the poor outcome in our study could be that 14 of 19 patients were classified as Lucarelli class 2 or 3, had high pre-transplant ferritin, and were heavily transfused. All of our patients' genotypes were beta thalassemia (homozygous), while the majority of patients in the study reported by Anurathapan et al. had β-thalassemia/hemoglobin E genotypes.
Conclusion
T cell replete haplo-HSCT is a feasible option for patients with no available HLA-matched donors. By expanding the donor pool to include haploidentical donors, we have increased the percentage of TM and SCD patients who may benefit from HSCT. Long-term follow-up is required for a better understanding of outcomes for these patients. This seems particularly true in developing countries where scarcity of resources poses major challenges.
Acknowledgements
Dr. Meet Kumar for statistical analysis.
Conflicts of Interest
The authors declare no conflict of interest.Disclosure forms provided by the authors are available here.
References
1. Lucarelli G, Gaziev J. Advances in the allogeneic transplantation for thalassemia. Blood Rev. 2008; 22: 53-63.
2. Shenoy S, Eapen M, Panepinto JA, Logan BR, Wu J, Abraham A, et al. A trial of unrelated donor marrow transplantation for children with severe sickle cell disease. Blood. 2016; 128: 2561-7.
3. Gragert L, Eapen M, Williams E, Freeman J, Spellman S, Baitty R, et al. HLA match likelihoods for hematopoietic stem-cell grafts in the U.S. registry. N Engl J Med. 2014; 371: 339-48.
4. Robinson TM, O'Donnell PV, Fuchs EJ, Luznik L. Haploidentical bone marrow and stem cell transplantation: experience with post-transplantation cyclophosphamide. Semin Hematol. 2016; 53: 90-7.
5. Luznik L, O'Donnell PV, Symons HJ, Chen AR, Leffell MS, Zahurak M, et al. HLA-Haploidentical Bone Marrow Transplantation for Hematologic Malignancies Using Nonmyeloablative Conditioning and High Dose, Post-transplantation Cyclophosphamide. Biol Blood Marrow Transplant. 2008; 14: 641-50.
6. Sodani P, Isgro A, Gaziev J, Polchi P, Paciaroni K, Marziali M, et al. Purified T-depleted, CD34+ peripheral blood and bone marrow cell transplantation from haploidentical mother to child with thalassemia. Blood. 2010; 115: 1296-302.
7. Isgro A, Marziali M, Sodani P, Gaziev J, Erer B, Polchi P, et al. Immunohematologic reconstitution in pediatric patients after T cell-depleted HLA-haploidentical stem cell transplantation for thalassemia. Biol Blood Marrow Transplant. 2010; 16: 1557-66.
8. Tolar J, Sodani P, Symons H. Alternative donor transplant of benign primary hematologic disorders. Bone Marrow Transplant. 2015; 50: 619-27.
9. Brissot E, Labopin M, Ehninger G, Stelljes M, Brecht A, Ganser A, et al. Haploidentical versus unrelated allogeneic stem cell transplantation for relapsed/refractory acute myeloid leukemia: a report on 1578 patients from the Acute Leukemia Working Party of the EBMT. Haematologica. 2019; 104: 524-32.
10. Anurathapan U, Hongeng S, Hematopoietic Stem Cell Transplantation for Homozygous β Thalassemia and β Thalassemia/ Hemoglobin E Patients from Haploidentical Donors. Bone Marrow Transplant. 2016; 51: 813-8.
11. Mahadeo KM, McArthur J, Adams RH, Radhi M, Angelo J, Jeyapalan A, et al. Consensus Report by the Pediatric Acute Lung Injury and Sepsis Investigators and Pediatric Blood and Marrow Transplant Consortium Joint Working Committees on Supportive Care Guidelines for Management of Veno-Occlusive Disease in Children and Adolescents: Part 2-Focus on Ascites, Fluid and Electrolytes, Renal, and Transfusion Issues. Biol Blood Marrow Transplant. 2017; 23: 2023-33.
12. Mohty M, Malard F, Abecassis M, Aerts E, Alaskar A S, Aljurf M_E, et al. Revised diagnosis and severity criteria for sinusoidal obstruction syndrome/veno-occlusive disease in adult patients: a new classification from the European Society for Blood and Marrow Transplantation. Bone Marrow Transplantation 2016; 51: 906-12.
13. Glucksberg H, Storb R, Fefer A, Buckner CD, Neiman PE, Clift RA, et al. Clinical manifestations of graft-versus-host disease in human recipients of marrow from HL-A-matched sibling donors. Transplantation 1974; 18: 295-304.
14. Bernaudin F, Socié G, Kuentz M, Chevret S, Duval M, Bertrand Y, et al. Long-term results of related myeloablative stem-cell transplantation to cure sickle cell disease. Blood. 2007; 110: 2749-56.
15. Caocci G, Orofino MG, Vacca A, Piroddi A, Piras E, Addari MC, et al. Long-term survival of beta thalassemia major patients treated with hematopoietic stem cell transplantation compared with survival with conventional treatment. Am J Hematol. 2017; 92: 1303-10.
16. Gaziev D, Galimberti M, Lucarelli G, Polchi P, Giardini C, Angelucci E, et al, Bone marrow transplantation from alternative donors for thalassemia: HLA-phenotypically identical relative and HLA-nonidentical sibling or parent transplants. Bone Marrow Transplantation. 2000; 25: 815-21.
17. Bolanos-Meade J, Fuchs EJ, Luznik L, Lanzkron SM, Gamper CJ, Jones RJ, et al. HLA-haploidentical bone marrow transplantation with post transplant cyclophosphamide expands the donor pool for patients with sickle cell disease. Blood. 2012; 120: 4285-91.
18. Bolanos-Meade J, Gamper C, Cooke KR, Jones RJ, Brodsky RA. Curative Allogeneic Bone Marrow Transplantation (AlloBMT) for Severe Hemoglobinopathies No Longer Requires Matched Donors or the Ability to Tolerate Myeloablative Conditioning. Abstract LBA3, Presented at BMT Tandem Meetings; Feb.21-25, 2018; Salt Lake City. https: //bmt.confex.com/tandem/2018/meetingapp.cgi/Paper/11769[Accessed September 1, 2020]
19. de la Fuente J, Dhedin N, Koyama T, Bernaudin F, Kuentz M, Karnik L, et al. Haploidentical Bone Marrow Transplantation with PostTransplantation Cyclophosphamide Plus Thiotepa Improves Donor Engraftment in Patients with Sickle Cell Anemia: Results of an International Learning Collaborative. Biol Blood Marrow Transplant. 2019; 25: 1197-1209.
20. Gaziev J, Isgrò A, Sodani P, Paciaroni K, et al. Haploidentical HSCT for hemoglobinopathies: improved outcomes with TCR αβ+/CD19+ depleted grafts. Blood Adv. 2018; 2: 263-70.
Search
News


