Volume 2 (2019) Issue 4 No.3 Pages 58-67
Abstract
Haematopoietic stem cell transplantation (HSCT) is now an established practice with over 70,000 transplants performed annually, and over 1.5 million around the world so far. The practice of HSCT has improved over the years due to advances in conditioning regiments, preparatory practices for patients leading up to the transplant, graft versus host disease (GVHD) and infection prophylaxis, as well as a better selection of patients. However, in many instances, the stem cells supplied to the patient may not be adequate for optimal transplantation outcomes. This may be seen in a few areas including umbilical cord blood transplantation, inadequate bone marrow, peripheral blood stem cell harvest, or gene therapy. Growing and expanding HSCs in culture would provide an increase in cell numbers prior to stem cell infusion and accelerate haematopoietic recovery, resulting in improved outcomes. Several new technologies have emerged in recent years, which have facilitated the expansion of haematopoietic stem and progenitor cells (HSPCs) in culture with good outcomes in vitro, in vivo, and in clinical trials. In this review, we will outline some of the reasons for the expansion of HSPCs as well as the new technologies facilitating the advances in HSCT
Evolution of haematopoietic stem cell transplantation (HSCT)
Haematopoietic stem cell transplantation (HSCT) is now an established clinical practice with over 70,000 transplants performed annually, and over 1.5 million around the world so far1. Since the first HSCT was carried out in identical twins2,3, several important milestones that allow HSCT to be carried out across international borders and between unrelated individuals4 have been achieved. These include the identification of the human leukocyte antigen (HLA) system5,6; development of effective immunosuppressive and anti-microbial drugs7; discovery of stem cell mobilizing agents8; and establishment of donor registries9. In recent years, HSCT has been enhanced by the advent of reduced intensity conditioning regimens especially for elderly patients (>55 years)10; innovative graft manipulations to mitigate graft-versushost-disease (GVHD) and maximize graft-versus-leukaemia (GVL)11; and advanced anti-microbial cellular therapeutics to provide effective infection control12. In addition, the use of donor lymphocyte infusion (DLI) to treat minimal residual disease (MRD) and post-transplant relapses13 along with an improved algorithm for patient selection14 has contributed to the advances in the use of HSCT15.
Amongst other factors, the outcome of HSCT strongly correlates with the quality and quantity of the hematopoietic stem and progenitor cells (HSPCs) that are delivered to the recipient16,17. In many instances, there could be inadequate number of HSPCs or HSPCs that are of suboptimal quality being supplied to the patients, resulting in poor outcomes measured through parameters such as neutrophil recovery and transplant-related mortality (TRM)18,19. Growing and expanding HSPCs in culture increase the number of desired potent cell populations (for example, cells that express surface markers such as CD34 and CD90) prior to infusion, which could potentially accelerate haematopoietic recovery and result in improved outcomes16,17. Several new technologies have emerged in recent years, which allow the expansion of primitive HSPCs in culture as demonstrated through effective in vitro phenotypic and in vivo functional assays followed by outstanding outcomes in pivotal early-phase clinical trials16-20. In this review, we will outline the major reasons for HSPC expansion as well as describe a selected group of new technologies facilitating the advances in HSCT and that will serve as a backbone of the new cellular therapy-based industry.
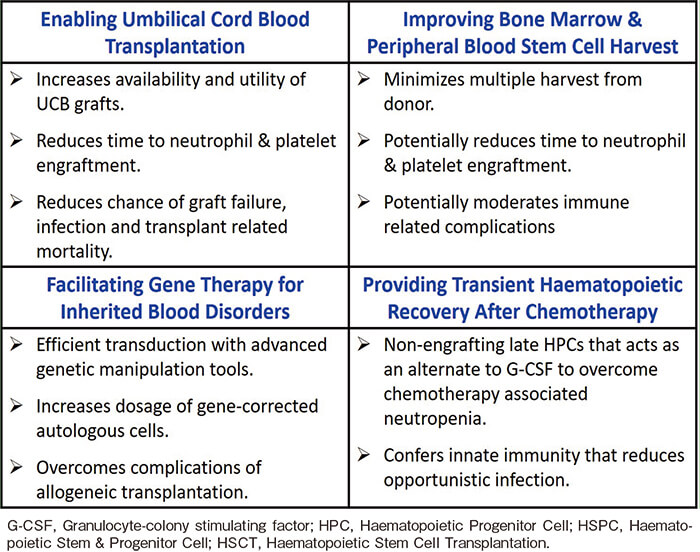
Clinical relevance of expanding HSPC to enable next-generation HSCT
Enabling umbilical cord blood (UCB) transplantation (UCBT)
UCB is now an FDA-approved HSPC source for HSCT21. The strong stem cell potential and immune pliability of UCB cells facilitate their use in HSCT despite requiring lower starting cell numbers than bone marrow (BM)or mobilized peripheral blood (mPB)along with the ability to cross histocompatibility barriers without in vitro or in vivo immune manipulation18,19. The immune naivety of UCB facilitates mismatched donor HSCT without an increase in GVHD and with retained GVT effects22,23. UCBT has been able to achieve equivalent outcomes, despite having one to two antigen mismatches when compared to fully matched BM or PB stem cell from unrelated donor24. Furthermore, a study conducted by Milano et al. showed that UCBT had better outcomes than bone marrow transplants (BMT) or mobilized peripheral blood stem cell transplants (PBSCT) due to a decrease in the relapse rates25. However, UCBT is fraught with the problem of inadequate or suboptimal cell dosage for transplantation as over 70% of the over 700,000 publicly banked cryo-preserved grafts fail to meet the minimal total nucleated cell (TNC) dosage of 25 million cells/kg of recipient?s body weight for most adults16,17. When UCBT is carried out with suboptimal cell dosage, TRM could exceed 30% but when a sufficient cell dose of UCB graft is infused, TRM could fall below 10%26. The infusion of a UCB graft with suboptimal cell dose causes delayed recovery of neutrophils and platelets, resulting in an increased risk of infections and bleeding compared to BMT or PBSCT27. Even with an optimal cell dose in a single or dual graft approach, neutrophil and platelet engraftment is slower compared to BMT or PBSCT by at least 1-2 weeks, increasing the chances of contracting life-threatening infection along with prolonged period of hospitalization28. The advent of more recent technologies in UCB expansion has resulted in accelerated haematopoietic recovery, with neutrophil engraftment being reduced from a median of 27 days to 11 days post-transplant16-20. Several other studies have also shown promising results that represent a significant potential advancement for UCBT which could facilitate its use as an important stem cell source to benefit at least more than 20,000 patients per annum worldwide16-20.
Improving BM and PBSC harvest
Inadequate HSPC dosage is a barrier to clinical usage of not only UCB grafts18,19; but also BM and mPB stem cell products which may fail to meet the optimal stem cell collection criteria to facilitate a standard HSCT29,30. BM and PBSC transplantations involve the harvest of HSPCs from a patient (autologous transplants) or from a donor (allogeneic transplants)31. BM harvests typically target 2-3×108 nucleated cells/kg of recipient bodyweight, and BM are extracted through multiple punctures carried out in the operating theatre for the extraction of approximately 1L of BM29. PBSC harvests are conducted after the injection of a HSPC mobilising agent such as G-CSF or plerixafor, followed by the collection of the cells through apheresis32,33. These harvests usually result in an adequate number of HSPC for transplantation; however, in many instances, stem cell harvests may not be adequate for the best transplant outcomes. For example, a recent study for mPB-based autologous HSCT demonstrated that a 5-day long subcutaneous administration of 15μg/kg of filgrastim resulted in 17%, 78%, and 5% of patients(n=102), achieving optimal (?5.0×106cells/kg), sub-optimal (?2.0-<5.0×106cells/kg), and poor (<2.0×106cells/kg) stem cell harvest for subsequent transplantation34,35. In allogeneic transplants, the problem of suboptimal cell dose of BM or mPB graft occurs particularly when the full or half HLA-matched donor: (1) is of a much smaller body size than the patient; (2) is of an older age than the patient; and (3) may have difficult venous access or some other health conditions. Autologous transplants patients, who are subjected to strong myelotoxic therapies to treat the primary disease preceding the stem cell harvest, are most likely to have insufficient collection of HSPC for subsequent infusion36. HSPC expansion could help us overcome the problem of inadequate stem cell harvest and, if established, may facilitate successful HSCT in these situations without inflicting unnecessary and excessive repeated harvests on the donor or patient. Furthermore, a reduced time required for haematopoietic recovery was observed with expanded UCB as described in subsequent sections; therefore, a similar expansion of BM or mPB grafts could further reduce the time to neutrophil engraftment from a median of 16 days to approximately 7 days (equivalent drop of 9 days when comparing median time to neutrophil engraftment of non-expanded versus expanded UCB- 25 days versus 13 days), thus enabling safer outpatient HSCT procedures with minimal post-transplant neutropenia16-20.
Facilitating gene therapy (GT) for inherited blood disorders
GT allows the correction of genetic disorders in haematopoietic cells and the treatment of patients with otherwise fatal or debilitating haematopoietic disorders such as thalassemia37 and sickle cell disease38. Despite the extensive amount of scientific studies that have been carried out in the field of GT over the past few decades, it has seen minimal clinical progress primarily due to problems such as (1) leukemogenesis caused by viral integration into unexpected sites39,40; and (2) poor transduction efficiency resulting in low number of transduced gene-corrected cells that does not meet the criteria of minimal cell dosage to carry out a successful HSCT41. However, in recent years, the regulatory approval and marketing of genetically engineered chimeric antigen receptor (CAR) T-cells primarily to treat CD19+diseases such as acute lymphoblastic leukaemia (ALL)42,43 and diffuse large B-cell lymphoma44 has re-ignited the hope of establishing GT as a clinical practice to improve patients’ lives. Newly established gene editing technologies using evolved and safer lenti-viral vectors45; clustered regularly interspaced short palindromic repeats (CRISPR) /CRISPR-associated protein 9 (CRISPR/Cas9)46; zinc finger nucleases47; and transcription activator-like effector nucleases48 could allow targeting and integration of specific gene sequences at precise locations of the genome, thus overcoming the problem of unexpected site integration and associated oncogenicity49. Ex vivo expansion of the gene-corrected HSPCs could facilitate the development of HSCT grafts that have sufficient therapeutic cell dosage of the genecorrected healthy cells to enable a successful transplantation through engraftment and subsequent reconstitution of the healthy blood cells.
Providing transient haematopoietic recovery after chemotherapy
Patients suffering from chemotherapy-associated neutropenia could also benefit from the infusion of late hematopoietic progenitor cells (HPCs) to minimize neutropenia and its associated complications through transient increase in infection-fighting white blood cells (WBCs)50. While most chemotherapy results in mild or negligible neutropenia, there are some, for example, induction chemotherapy for acute myeloid leukaemia (AML) that may result in profound neutropenia for more than 2 weeks51. Furthermore, cytotoxic antineoplastic therapy administered as conditioning regimen prior to HSCT adversely impacts both myelopoiesis and the integrity of the gastrointestinal mucosa, thus exponentially increasing the risk of life-threatening infections in these patients who are unable to mount an inflammatory response52. Current clinical management of high-risk chemotherapy-associated neutropenia (absolute neutrophil count, ANC<500cells/μL for>7 days) and its associated opportunistic infections involve prophylactic mono- or combination therapy of antimicrobial drug/s along with recombinant human hematopoietic growth factors such as G-CSF to stimulate the production of neutrophils53,54. Although G-CSF has shown proven efficacy in shortening the period of chemotherapy-induced neutropenia, it is ineffective in very early HSPCs but only effective in committed/late hematopoietic progenitor cells (HPCs) once the BM has partially recovered55. Based on epidemiological studies carried out by a pharmaceutical company (Pfizer, Bristol-Myers Squibb, Novartis), the number of HSCTs performed is expected to rise worldwide in the next decade or so due to the increasing incidence of leukaemia and other BM disorders, improved transplant outcomes, and growth in the number of transplant centres and donor registries especially in densely populated regions of Asia. To reduce post-transplant complication rates associated with the rising trend, there is an urgent need to develop therapeutic strategies to mitigate neutropenia that occurs after chemotherapy and during the pre-engraftment phase following HSCT. Because the expansion and infusion of late/committed HPCs, preferably obtained from on-demand, off-the-shelf, and non-HLA matched donors, do not retain the ability to impart life-long haematopoiesis and could only provide a wave of early and late myeloid cells to impart innate immunity while awaiting for the patient’ s endogenous BM recovery, they act as a lucrative alternative to recombinant G-CSF therapy. Such non-engrafting expanded HSCT grafts that could tackle neutropenia could potentially have applicability in areas of solid tumours and organ transplantation, and they are being developed by several groups56 (Table 1).
Methods for expanding early HSPCs and late HPCs to provide long-term and transient hematopoiesis
Majority of the HSPC expansion studies were performed using UCB grafts given that its clinical usage is primarily limited by low cell dosage16-20. Over the past two decades, up to 15 different clinical trials have explored various methods of expanding UCB HSPC which have resulted in the recruitment of over 350 patients57. All ex vivo HSPC expansion cultures involve the use of various early-acting cytokines such as stem cell factor (SCF), thrombopoietin (TPO), and Flt-3 ligand (Flt-3L)for maintaining stemness, whilst late-acting cytokines such as interleukin (IL) -3, IL-6, IL-11, and granulocyte-colony stimulating factor (G-CSF) are added to support rapid proliferation accompanied by differentiation16-20. In recent years, a number of novel growth factors such as Notch ligand58, insulin-like growth factorbinding protein-2 (IGFBP-2)59, angiopoietin-like (Angptl) proteins60 and pleiotrophin61 have been discovered and used in various expansion technologies. In addition to a cytokine cocktail, clinical trials have involved the use of mesenchymal stromal cell (MSC) co-culture system that mimics the BM niche to expand non-enriched UCB grafts62 and bio-reactors to enable up-scaled culturing of CD34-selected HSPCs63. Furthermore, they have involved the addition of several small molecules associated with HSPC maintenance and proliferation such as tetraethylenepentamine (copper chelator that modulates differentiation)64,65, nicotinamide (sirtuin-1 inhibitors)66,67, StemRegenin-1 (aryl hydrocarbon receptor antagonist)68, and UM171 (mechanism of action not known)69. Recent pre-clinical studies have demonstrated the roles of a range of new small molecules in the expansion of HSPCs. They include P18IN003, P18IN011, and XIE18-6 (cyclin-dependent kinase inhibitors)70; NR101 (non-peptidyl small molecule agonist of c-MPL)71; eltrombopag (a human specific thrombopoietin receptor agonist)72; CHIR99021 and rapamycin (Wnt and β catenin pathway modulators)73; and 5-azacytidine, trichostatin A, garcinol, and valproic acid (epigenetic modulators)74,75. Others such as resveratrol (a naturally occurring polyphenol)76; serotonin (a monoamine neurotransmitter)77; GW9662 (a PPARγ antagonist)78; SB203580, Vx702, BIRB-796 and Ly2228820 (potent inhibitors of p38 mitogen-activated protein kinases)79; and C7 (a new structural analogue of SB203580)80 have also been used in HSPC expansion studies. Amongst these compounds, only C7 has been shown to be able to expand HSPC from both non-enriched and CD34- enriched grafts that retain both in vitro primitive HSPC phenotype and long-term in vivo functionality in xenotransplantation model80. While some of the methods have met with limited success, we would like to highlight a few that have had effective clinical outcomes in recent years and could potentially change the field of HSCT.
Omidubicel/NiCord®, MGTA-456, and ECT-001 expanded HSPC grafts to enable UCBT
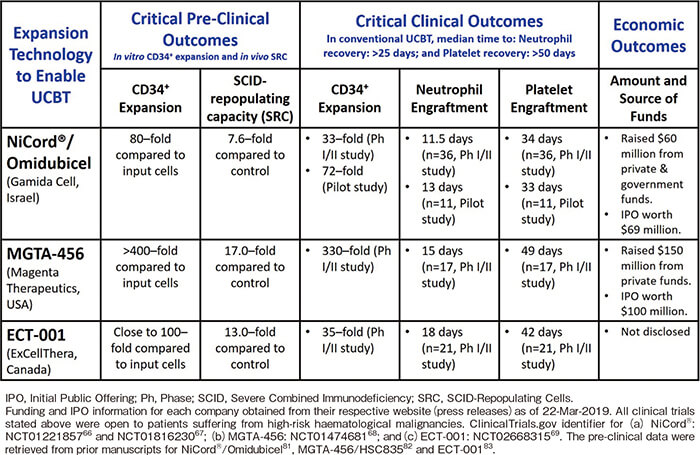
Gamida Cell (Israel) has established nicotinamide (NAM), a form of vitamin B3, as a novel agent to expand CD34/CD133 selected UCB cells81, and the manufactured cell therapy product is referred to as Omidubicel/NiCord®. High-throughput screening of over 100,000 heterocycles compounds identified a purine derivative, StemRegenin-1 (SR-1), that could increase the absolute number of CD34+ cells by inhibiting aryl hydrocarbon receptor in ex vivo expansion cultures82 resulting in an investigational product, MGTA-456/HSC83568, by Novartis (Switzerland) and Magenta Therapeutics (MA, USA). Similarly, another manipulated UCB graft known as ECT-001 was developed by ExCellThera (Canada) using UM171, a pyrimidoindole derivative83 discovered via high-throughput screening of over 5,300 low molecular weight compounds.
Omidubicel (single centre pilot study, n=11, with ClinicalTrials.gov identifier: NCT0122185766 and multicentre phase?/? study, n=36, with ClinicalTrials.gov identifier: NCT0181623067), MGTA-456 (single centre pilot study, n=17, with ClinicalTrials.gov identifier: NCT0147468168), and ECT-001 (single centre pilot study forn=21 with Clinical Trials . govidentifier :NCT0266831569) underwent clinical trial for patients (total n=85) suffering from high-risk haematological malignancies who received myeloablative conditioning regimen and standard GvHD prophylaxis. The production process for Omidubicel requires up to 27 days and involves the culturing of CD133+/CD34+UCB cells in Minimum Essential Medium α supplemented with 10% FBS, 50 ng/mL of SCF, 50 ng/mL of TPO, 50 ng/mL of Flt-3L, 50 ng/mL of IL-6 and 2.5 mM of NAM66,67. Compared to Omidubicel, the manufacturing process of MGTA-456 takes a shorter duration of up to 14 days and involves the culturing of the CD34+ UCB cells in serumfree expansion media supplemented with 50 ng/mL of SCF, 50 ng/mL of TPO, 50 ng/mL of Flt-3L, 50 ng/mL of IL-6, and 750 nM of SR-168. ECT-001 manufacturing requires only 7 days and involves the culturing of purified CD34+ UCB cells using a fed-batch closed bioreactor with serum-free expansion medium supplemented with 100 ng/mL of SCF and 100 ng/mL of Flt-3L, 50 ng/mL of TPO, 10μg/mL of low density lipo-proteins, and 35-50 nM of UM17169. The CD133–/CD34– fraction of Omidubicel, MGTA-456, and ECT-001 were cryopreserved for infusion into the patients along with the manipulated CD34+ fraction thus enabling the infusion of minimally manipulated T, B, and other immune cells that potentially supports engraftment after transplantation66-69. Production failure rates of approximately 10% and 22% was reported for Omidubicel and MGTA-456, respectively66-68. Majority of the patients (71%) in these trials received a second unmanipulated UCB graft along with expanded UCB product as a measure of clinical safety.
All the stated clinical trials reported significant expansion of CD34 cells (as outlined in Table 2) which resulted in patients receiving significantly higher cell dose (TNC and CD34) that resulted in faster hematopoietic recovery66-69 for graft-derived neutrophils and platelets (Table 2). Post-transplant acute GvHD (aGvHD) for Omidubicel recipients manifested primarily as grade ? to ? whereas MGTA-456 and ECT-001 exhibited very low risk of aGvHD66-69. Immune cell reconstitution of patients receiving these investigational cell therapy products did not exhibit any abnormalities compared to conventional UCBT66-69. Transplantation of Omidubicel, MGTA-456, and ECT-001 did not cause any acute infusional toxicities (<24 hours) or adverse events (up to 30 days posttransplant), and no patient experienced graft failure66-69. Furthermore, both in vitro experiments (telomere length, proliferation rates, and pathology-related tests including cytogenetics)82-83 and long-term follow up of patients66-69 enrolled in these studies did not indicate any possible mutagenesis or leukemogenesis of the culture expanded CD34 cells. A major advantage of Omidubicel, MGTA- 456, and ECT-001 recipients manifested in a shorter hospital stay by at least 2 weeks compared to historical patients receiving conventional single or double UCBT66-69 suggesting a significant drop in hospitalization associated costs. Another advantage of ex vivo HSPC expansion as demonstrated by the ECT-001 study is that it allowed the selection of UCB units with a better HLAmatching rather than with a higher cell dosage69 which resulted in the clinical team having access to almost half of the UCBs in the banks instead of the typical 5% that is available for a standard 70 kg patient69.
Currently Omidubicel is being evaluated in a multicentre, randomized, phase? registration trial (Clinical- Trials.gov identifier: NCT02730299) that aims to recruit close to 120 patients suffering from haematological malignancies. Stand-alone MGTA-456 graft is being evaluated in ongoing phase? studies for patients suffering from haematological malignancies (ClinicalTrials.gov identifier: NCT03674411) and inherited metabolic disorders (ClinicalTrials.gov identifier: NCT03406962) with plans to extend its application to the treatment of sickle cell anaemia. Similarly, the clinical application for ECT- 001 is being broadened through a single centre phase?/ ? trial (ClinicalTrials.gov identifier: NCT03441958) involving high risk multiple myeloma patients.
NLA101-Notch Ligand-based expansion of late HPC by NOHLA Therapeutics (USA) to overcome prolonged chemotherapy-associated neutropenia
Initial laboratory studies demonstrated the role of Notch signalling pathway in controlling the fate of CD34+ HSPC, for example, the overexpression of Notch-1 gene in CD34+ cells allowed enhanced selfrenewal capacity84. The NLA-101 expansion process by NOHLA Therapeutics (CA, USA) requires up to 16 days where purified UCB CD34+ cells (from frozen or fresh grafts) are cultured in serum-free expansion medium supplemented with 300 ng/mL of SCF, 300 ng/mL of FLT-3L, 100 ng/mL of TPO, 100 ng/mL of IL-6, 10 ng/ mL of IL-3, and the required density of Delta 1 (Ext-IgG)56,85. The CD34– fraction containing the lymphoid cells is discarded and not infused into the patient unlike Omidubicel, MGTA-456, and ECT-00166-69. In an early phase pilot study (ClinicalTrials.gov identifier:NCT00343798), NLA-101 was administered to ten myeloablated patients suffering from haematological malignancies along with a second unmanipulated graft85. The expansion process resulted in 164-fold expansion of the absolute CD34+ cell dosage and resulted in a faster median time to neutrophil recovery at 16 days compared to 26 days in the control cohort85. Majority of the patients exhibited the engraftment of NLA-101 at early time points, whereas only a couple of the patients retained long-term hematopoiesis from the expanded graft while the rest had stable engraftment from the second unmanipulated graft. Although NLA-101 proved to be safe, with no adverse outcomes with regards to graft failure, GvHD or relapse but non-persistence of the expanded unit raised concerns that the ex vivo expansion process was depleting true HSPCs or could be producing cells that merely facilitated the engraftment of the unmanipulated UCB graft85.
Based on the outcomes of the pilot clinical study, the clinical use of NLA-101 was repurposed as an off-theshelf (i.e. no matching on HLA is required) cellular therapy product to overcome neutropenia and reduce infections in patients receiving high-intensity chemotherapy56. Twenty-nine patients suffering from AML were recruited for a phase? study (ClinicalTrials.gov identifier: NCT01031368) as infection is a major cause of posttreatment mortality and morbidity56. The UCB grafts that underwent expansion resulted in a median expansion of 129- and 973-fold for CD34+ cells and TNC, respectively56. Patients received NLA-101 at the end of the first induction cycle and were eligible for a second infusion of NLA-101 with consolidation therapy if they lacked residual leukaemia, toxicity associated with first NLA-101 infusion, or uncontrolled infections56. GvHD was not observed in any patient and routine chimerism analysis showed that NLA-101 provided transient engraftment up to 14 days post-infusion56. The median time to autologous neutrophil and platelet recovery were 19 and 27 days, respectively, which were similar to control cohort56. In conclusion, the study demonstrated that AML patients receiving NLA-101 had a decrease in infection incidence and the time to infection without any noted adverse outcomes56.
Conclusion
HSPC expansion, particularly from UCB, is now a reality. The impact on the efficacy, safety, and utilisation of UCB for HSCT cannot be underestimated. Improving the safety and results of expanded UCBT would encourage more physicians to perform UCBT for their patients. Furthermore, with the potential significant reduction in hospitalisation, more centres would be keen to adopt this approach. The cost reduction in hospitalisation could also be attractive to insurance companies and other payers even if there are some costs associated with the ex vivo CD34+ expansion process. The use of HSPC expansion platforms to mitigate the problem of inadequate harvest of BM or mPB would also significantly increase the market for the expanded HSPC products. Whilst clinical trials are still underway, the use of expanded HSCT grafts in post-chemotherapy or GT settings could further increase the impact of HSCT.
Acknowledgments
The authors would like to acknowledge National Health Innovation Centre Singapore for translational funding support (NHIC Ref: NHIC-I2D-1506047). Laboratory space and research administrative support were provided by National Cancer Centre Singapore and Duke-NUS Medical School, Singapore.
Authors’ Contributions
William YK Hwang conceived, reviewed, and revised the manuscript; Paul Chong transcribed the original draft and added new text and references; Sudipto Bari contributed substantially to the revision of the manuscript.
Conflict of Interest
William YK Hwang and Sudipto Bari have patents in hematopoietic stem cell expansion using small moleculebased platform. Disclosure forms provided by the authors are available here.
References
1. Gratwohl A, Baldomero H, Aljurf M, Pasquini MC, Bouzas LF & Yoshimi A, et al. Hematopoietic stem cell transplantation: a global perspective. JAMA. 2010; 303: 1617-24.
2. Little M, Storb R. History of haematopoietic stem-cell transplantation. Nat Rev Cancer. 2002; 2: 231-8.
3. Thomas ED, Blume KG. Historical markers in the development of allogeneic hematopoietic cell transplantation. Biol Blood Marrow Transplant. 1999; 5: 341-36.
4. Juric MK, Ghimire S, Ogonek J, Weissinger EM, Holler E, van Rood JJ, et al. Milestones of Hematopoietic Stem Cell Transplantation- From First Human Studies to Current Developments. Front Immunol. 2016; 7: 470.
5. Nowak J. Role of HLA in hematopoietic SCT. Bone Marrow Transplant. 2008; 42 (Suppl 2): S71-6.
6. Fürst, D, Müller C, Vucinic V, Bunjes D, Herr W, Gramatzki M, et al. High-resolution HLA matching in hematopoietic stem cell transplantation: a retrospective collaborative analysis. Blood. 2013; 122: 3220-9.
7. Craddock C. Haemopoietic stem-cell transplantation: recent progress and future promise. Lancet Oncol. 2000; 1: 227-34.
8. Dale DC. The discovery, development and clinical applications of granulocyte colony-stimulating factor. Trans Am Clin Climatol Assoc. 1998; 109: 27-36.
9. Petersdorf EW. The World Marrow Donor Association: 20 years of international collaboration for the support of unrelated donor and cord blood hematopoietic cell transplantation. Bone Marrow Transplant. 2010; 45: 807-10.
10. Gyurkocza B, Sandmaier BM. Conditioning regimens for hematopoietic cell transplantation: one size does not fit all. Blood. 2014; 124: 344-53.
11. Triplett BM, Shook DR, Eldridge P, Li Y, Kang G, Dallas M, et al. Rapid memory T-cell reconstitution recapitulating CD45RA-depleted haploidentical transplant graft content in patients with hematologic malignancies. Bone Marrow Transplant. 2015; 50: 968-77.
12. Leen AM, Bollard CM, Mendizabal AM, Shpall EJ, Szabolcs P, Antin JH, et al. Multicenter study of banked third-party virusspecific T cells to treat severe viral infections after hematopoietic stem cell transplantation. Blood. 2013; 121: 5113-23.
13. Dominietto A, Pozzi S, Miglino M, Albarracin F, Piaggio G, Bertolotti F, et al. Donor lymphocyte infusions for the treatment of minimal residual disease in acute leukemia. Blood. 2007; 109: 5063-504.
14. Norkin M, Wingard JR. Recent advances in hematopoietic stem cell transplantation. Version 1. F1000Res. 2017; 6: 870.
15. Jeng RR, van den Brink MR. Allogeneic haematopoietic stem cell transplantation: individualized stem cell and immune therapy of cancer. Nat Rev Cancer. 2010; 10: 213-21.
16. Lund TC, Boitano AE, Delaney CS, Shpall EJ & Wagner JE. Advances in umbilical cord blood manipulation-from niche to bedside. Nat Rev Clin Oncol. 2015; 12: 163-74.
17. Bari S, Seah KKH, Poon Z, Cheung AMS, Fan X, Ong SY, et al. Expansion and Homing of Umbilical Cord Blood Hematopoietic Stem and Progenitor Cells for Clinical Transplantation. Biol Blood Marrow Transplant. 2015; 21: 1008-19.
18. Ballen KK, Gluckman E, Broxmeyer HE. Umbilical cord blood transplantation: the first 25 years and beyond. Blood. 2013; 122: 491-8.
19. Norkin M, Lazarus HM, Wingard JR. Umbilical cord blood graft enhancement strategies: has the time come to move these into the clinic? Bone Marrow Transplant. 2013; 48: 884-9.
20. Horwitz ME, Frassoni F. Improving the outcome of umbilical cord blood transplantation through ex vivo expansion or graft manipulation. Cytotherapy. 2015; 17: 730-8.
21. Voelker R. FDA grants approval for first cord blood product. JAMA. 2011; 306: 2442.
22. Rocha V, Wagner JE, Sobocinski KA, Klein JP, Zhang MJ, Horowitz MM, Gluckman E. Graft-versus-host disease in children who have received a cord-blood or bone marrow transplant from an HLA-identical sibling. Eurocord and International Bone Marrow Transplant Registry Working Committee on Alternative Donor and Stem Cell Sources. N Engl J Med. 2000; 342: 1846-54.
23. Cunha R, Loiseau P, Ruggeri A, Sanz G, Michel G, Paolaiori A, et al. Impact of HLA mismatch direction on outcomes after umbilical cord blood transplantation for hematological malignant disorders: a retrospective Eurocord-EBMT analysis. Bone Marrow Transplant. 2014; 49: 24-9.
24. Hwang WYK, Samuel M, Tan D, Koh LP, Lim W, Linn YC. A meta-analysis of unrelated donor umbilical cord blood transplantation versus unrelated donor bone marrow transplantation in adult and pediatric patients. Biol Blood Marrow Transplant. 2007; 13: 444-53.
25. Milano F, Gooley T, Wood B, Woolfrey A, Flowers ME, Doney K, et al. Cord-Blood Transplantation in Patients with Minimal Residual Disease. N Engl J Med. 2016 8; 375: 944-53.
26. Michel G, Rocha V, Chevret S, Arcese W, Chan KW, Filipovich A, et al. Unrelated cord blood transplantation for childhood acute myeloid leukemia: a Eurocord Group analysis. Blood. 2003; 102: 4290-7.
27. Laughlin MJ, Eapen M, Rubinstein P, Wagner JE, Zhang MJ, Champlin RE, et al. Outcomes after transplantation of cord blood or bone marrow from unrelated donors in adults with leukemia. N Engl J Med. 2004; 351: 2265-75.
28. Sideri A, Neokleous N, Brunet De La Grange P, Guerton B, Le Bousse Kerdilles MC, Uzan G, Peste-Tsilimidos C, Gluckman E. An overview of the progress on double umbilical cord blood transplantation. Haematologica. 2011; 96: 1213-20.
29. Halter J, Kodera Y, Ispizua AU, Greinix HT, Schmitz N, Favre G, et al. Severe events in donors after allogeneic hematopoietic stem cell donation. Haematologica. 2009; 94: 94-101.
30. Anderlini P, Rizzo JD, Nugent ML, Schmitz N, Champlin RE, Horowitz MM; IBMTR Statistical Center of the International Bone Marrow Transplant Registry, Medical College of Wisconcin, Milwaukee, WI, USA; EBMT, Kiel, Germany. Peripheral blood stem cell donation: an analysis from the International Bone Marrow Transplant Registry (IBMTR) and European Group for Blood and Marrow Transplant (EBMT) databases. Bone Marrow Transplant. 2001; 27: 689-92.
31. Hequet O. Hematopoietic stem and progenitor cell harvesting: technical advances and clinical utility. J Blood Med. 2015; 6: 55-67.
32. Spoerl S, Peter R, Wäscher D, Götze K, Verbeek M, Peschel C, Krackhardt AM. Patients? outcome after rescue plerixafor administration for autologous stem cell mobilization: a singlecenter retrospective analysis. Transfusion. 2017; 57: 115-21.
33. Nademanee AP, DiPersio JF, Maziarz RT, Stadtmauer EA, Micallef IN, Stiff PJ, et al. Plerixafor plus granulocyte colonystimulating factor versus placebo plus granulocyte colonystimulating factor for mobilization of CD34 (+) hematopoietic stem cells in patients with multiple myeloma and low peripheral blood CD34 (+) cell count: results of a subset analysis of a randomized trial. Biol Blood Marrow Transplant. 2012; 18: 1564-72.
34. Osmani S, Brahimi M, Talhi S, Amani K, Ouldjeriouat H, Bouchama S, et al. A Comparison of Efficacy Following Two Different Doses of Granulocyte Colony Stimulating Factor (G-CSF) Alone for Mobilization of Peripheral Blood Stem Cells in 221 Multiple Myeloma Patients. Blood. 2016; 128: 5732.
35. Li J, Hamilton E, Vaughn L, Graiser M, Renfroe H, Lechowicz MJ, et al. Effectiveness and cost analysis of “just-intime” salvage plerixafor administration in autologous transplant patients with poor stem cell mobilization kinetics. Transfusion. 2011; 51: 2175-82.
36. Visram A, Bredeson C, Allan D, Sabloff M, Huebsch L, Tay J, et al. Long-term graft function following autologous hematopoietic cell transplantation and the impact of preemptive plerixafor in predicted poor mobilizers. Blood Cancer J. 2018; 8: 14.
37. Thompson AA, Walters MC, Kwiatkowski J, Rasko JEJ, Ribeil JA, Hongeng S, et al. Gene Therapy in Patients with Transfusion- Dependent β-Thalassemia. N Engl J Med. 2018; 378: 1479-93.
38. Rita Rubin MA. Gene Therapy for Sickle Cell Disease Shows Promise. JAMA. 2019; 321: 334.
39. Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008; 118: 3143-50.
40. Ruella M, Xu J, Barrett DM, Fraietta JA, Reich TJ, Ambrose DE, et al. Induction of resistance to chimeric antigen receptor T cell therapy by transduction of a single leukemic B cell. Nat Med. 2018; 24: 1499-503.
41. Chira S, Jackson CS, Oprea I, Ozturk F, Pepper MS, Diaconu I, et al. Progresses towards safe and efficient gene therapy vectors. Oncotarget. 2015; 6: 30675-703.
42. Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med. 2013; 368: 1509-18.
43. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014; 371: 1507-17.
44. Schuster SJ, Bishop MR, Tam CS, Waller EK, Borchmann P, McGuirk, JP, et al. Tisagenlecleucel in Adult Relapsed or Refractory Diffuse Large B-Cell Lymphoma. N Engl J Med. 2019; 380: 45-56.
45. Lundstrom K. Viral Vectors in Gene Therapy. Diseases. 2018; 6: 42.
46. Ratner HK, Sampson TR, Weiss DS. Overview of CRISPRCas9 Biology. Cold Spring Harb Protoc. 2016; 2016: 1023-38.
47. Klug A. The discovery of zinc fingers and their applications in gene regulation and genome manipulation. Annu Rev Biochem. 2010; 79: 213-31.
48. Joung JK, Sander JD. TALENs: a widely applicable technology for targeted genome editing. Nat Rev Mol Cell Biol. 2013; 14: 49-55.
49. Gaj T, Gersbach CA, Barbas CF. ZFN, TALEN, and CRISPR/ Cas-based methods for genome engineering. Trends Biotechnol. 2013; 31: 397-405.
50. BitMansour A, Cao TM, Chao S, Shashidhar S, Brown JM. Single infusion of myeloid progenitors reduces death from Aspergillus fumigatus following chemotherapy-induced neutropenia. Blood. 2005; 105: 3535-7.
51. Buckley SA, Othus M, Vainstein V, Abkowitz JL, Estey EH, Walter RB. Prediction of adverse events during intensive induction chemotherapy for acute myeloid leukemia or highgrade myelodysplastic syndromes. Am J Hematol. 2014; 89: 423-8.
52. Tavaré A, O?Flynn N. Recognition, diagnosis, and early management of sepsis: NICE guideline. Br J Gen Pract. 2017; 67: 185-6.
53. Maher DW, Lieschke GJ, Green M, Bishop J, Stuart-Harris R, Wolf M, et al. Filgrastim in patients with chemotherapyinduced febrile neutropenia. A double-blind, placebo-controlled trial. Ann Intern Med. 1994; 121: 492-501.
54. Ozkaynak MF, Krailo M, Chen Z, Feusner J. Randomized comparison of antibiotics with and without granulocyte colonystimulating factor in children with chemotherapy-induced febrile neutropenia: a report from the Children?s Oncology Group. Pediatr Blood Cancer. 2005; 45: 274-80.
55. Blair HA, Scott LJ. Tbo-Filgrastim: A Review in Neutropenic Conditions. BioDrugs. 2016; 30: 153-60.
56. Delaney C, Milano F, Cicconi L, Othus M, Becker PS, Sandhu V, et al. Infusion of a non-HLA-matched ex-vivo expanded cord blood progenitor cell product after intensive acute myeloid leukaemia chemotherapy: a phase 1 trial. Lancet Haematol. 2016; 3: e330-9.
57. Kiernan J, Damien P, Monaghan M, Shorr R, McIntyre L, Fergusson D, Tinmouth A, Allan D. Clinical Studies of Ex Vivo Expansion to Accelerate Engraftment After Umbilical Cord Blood Transplantation: A Systematic Review. Transfus Med Rev. 2017; 31: 173-82.
58. Lampreia FP, Carmelo JG, Anjos-Afonso F. Notch Signaling in the Regulation of Hematopoietic Stem Cell. Curr Stem Cell Rep. 2017; 3: 202-9.
59. Zhang CC, Kaba M, Iizuka S, Huynh H, Lodish HF. Angiopoietin- like 5 and IGFBP2 stimulate ex vivo expansion of human cord blood hematopoietic stem cells as assayed by NOD/SCID transplantation. Blood. 2008; 111: 3415-23.
60. Zhang CC, Kaba M, Ge G, Xie K, Tong W, Hug C, Lodish HF. Angiopoietin-like proteins stimulate ex vivo expansion of hematopoietic stem cells. Nat Med. 2006; 12: 240-5.
61. Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010; 16: 475-82.
62. de Lima M, McNiece I, Robinson SN, Munsell M, Eapen M, Horowitz M, et al. Cord-blood engraftment with ex vivo mesenchymal- cell coculture. N Engl J Med. 2012; 367: 2305-15.
63. Jaroscak J, Goltry K, Smith A, Waters-Pick B, Martin PL, Driscoll TA, et al. Augmentation of umbilical cord blood (UCB)transplantation with ex vivo-expanded UCB cells: results of a phase 1 trial using the AastromReplicell System. Blood. 2003; 101: 5061-7.
64. de Lima M, McMannis J, Gee A, Komanduri K, Couriel D, Andersson BS, et al. Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase?/? clinical trial. Bone Marrow Transplant. 2008; 41: 771-8.
65. Stiff PJ, Montesinos P, Peled T, Landau E, Goudsmid NR, Mandel J, et al. Cohort-Controlled Comparison of Umbilical Cord Blood Transplantation Using Carlecortemcel-L, a Single Progenitor-Enriched Cord Blood, to Double Cord Blood Unit Transplantation. Biol Blood Marrow Transplant. 2018; 24: 1463-70.
66. Horwitz ME, Chao NJ, Rizzieri DA, Long GD, Sullivan KM, Gasparetto C, Chute JP, et al. Umbilical cord blood expansion with nicotinamide provides long-term multilineage engraftment. J Clin Invest. 2014; 124: 3121-8.
67. Horwitz ME, Wease S, Blackwell B, Valcarcel D, Frassoni F, Boelens JJ, et al. Phase?/? Study of Stem-Cell Transplantation Using a Single Cord Blood Unit Expanded Ex Vivo With Nicotinamide. J Clin Oncol. 2019; 37: 367-74.
68. Wagner JE, Brunstein CG, Boitano AE, DeFor TE, McKenna D, Sumstad D, et al. Phase?/? Trial of StemRegenin-1 Expanded Umbilical Cord Blood Hematopoietic Stem Cells Supports Testing as a Stand-Alone Graft. Cell Stem Cell. 2016; 18: 144-55.
69. Cohen S, Roy J, Lachance S, Marinier A, Delisle JS, Roy DC, et al. Single UM171 Expanded Cord Blood Permits Transplantation of Better HLA Matched Cords with Excellent Gvhd Relapse Free Survival. Blood. 2018; 132: 4658.
70. Xie XQ, Yang P, Zhang Y, Zhang P, Wang L, Ding Y, et al. Discovery of novel INK4C small-molecule inhibitors to promote human and murine hematopoietic stem cell ex vivo expansion. Sci Rep. 2015; 5: 18115.
71. Nishino T, Miyaji K, Ishiwata N, Arai K, Yui M, Asai Y, et al. Ex vivo expansion of human hematopoietic stem cells by a smallmolecule agonist of c-MPL. Exp Hematol. 2009; 37: 1364-77.
72. Sun H, Tsai Y, Nowak I, Liesveld J, Chen Y. Eltrombopag, a thrombopoietin receptor agonist, enhances human umbilical cord blood hematopoietic stem/primitive progenitor cell expansion and promotes multi-lineage hematopoiesis. Stem Cell Res. 2012; 9: 77-86.
73. Huang J, Nguyen-McCarty M, Hexner EO, Danet-Desnoyers G, Klein PS. Maintenance of hematopoietic stem cells through regulation of Wnt and mTOR pathways. Nat Med. 2012; 18: 1778-85.
74. Mahmud N, Petro B, Baluchamy S, Li X, Taioli S, Lavelle D, et al. Differential effects of epigenetic modifiers on the expansion and maintenance of human cord blood stem/progenitor cells. Biol Blood Marrow Transplant. 2014; 20: 480-9.
75. Chaurasia P, Gajzer DC, Schaniel C, D?Souza S, Hoffman R. Epigenetic reprogramming induces the expansion of cord blood stem cells. J Clin Invest. 2014; 124: 2378-95.
76. Heinz N, Ehrnström B, Schambach A, Schwarzer A, Modlich U, Schiedlmeier, B. Comparison of Different Cytokine Conditions Reveals Resveratrol as a New Molecule for Ex Vivo Cultivation of Cord Blood-Derived Hematopoietic Stem Cells. Stem Cells Transl Med. 2015; 4: 1064-72.
77. Yang M, Li K, Ng PC, Chuen CK, Lau TK, Cheng YS, et al. Promoting effects of serotonin on hematopoiesis: ex vivo expansion of cord blood CD34+stem/progenitor cells, proliferation of bone marrow stromal cells, and antiapoptosis. Stem Cells. 2007; 25: 1800-6.
78. Guo B, Huang X, Lee MR, Lee SA, Broxmeyer HE. Antagonism of PPAR-γ signaling expands human hematopoietic stem and progenitor cells by enhancing glycolysis. Nat Med. 2018; 24: 360-7.
79. Baudet A, Karlsson C, Safaee Talkhoncheh M, Galeev R, Magnusson M, Larsson J. RNAi screen identifies MAPK14 as a druggable suppressor of human hematopoietic stem cell expansion. Blood. 2012 28; 119: 6255-8.
80. Bari S, Zhong Q, Fan X, Poon Z, Lim AST, Lim TH, et al. Ex Vivo Expansion of CD34+CD90+CD49f+Hematopoietic Stem and Progenitor Cells from Non-Enriched Umbilical Cord Blood with Azole Compounds. Stem Cells Transl Med. 2018; 7: 376-93.
81. Peled T, Shoham H, Aschengrau D, Yackoubov D, Frei G, Rosenheimer GN, et al. Nicotinamide, a SIRT1 inhibitor, inhibits differentiation and facilitates expansion of hematopoietic progenitor cells with enhanced bone marrow homing and engraftment. Exp Hematol. 2012; 40: 342-55.
82. Boitano AE, Wang J, Romeo R, Bouchez LC, Parker AE, Sutton SE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010; 329: 1345-8.
83. Fares I, Chagraoui J, Gareau Y, Gingras S, Ruel R, Mayotte N, et al. Cord blood expansion. Pyrimidoindole derivatives are agonists of human hematopoietic stem cell self-renewal. Science. 2014; 345: 1509-12.
84. Karanu FN, Murdoch B, Gallacher L, Wu DM, Koremoto M, Sakano S, Bhatia M. The notch ligand jagged-1 represents a novel growth factor of human hematopoietic stem cells. J Exp Med. 2000; 192: 1365-72.
85. Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med. 2010; 16: 232-6.
Search
News


