Volume 2 (2019) Issue 2 No.3 Pages 31-35
Abstract
Hematopoietic cell transplantation (HCT) confers a long-term disease-modifying therapy for transplant-permissive inherited metabolic diseases (IMDs). We examined the overall survival (OS) and engrafted survival (ES) of children with IMDs, who received first HCT at Royal Manchester Children?s hospital from 1985 to 2016. A total of 137 children with IMDs were included in this analysis (historical cohort [1985-2006], n=65; current cohort [2007-2016], n=72). Primary diagnoses included mucopolysaccharidoses (81%), X-linked adrenoleukodystrophy (6%), metachromatic leukodystrophy (4%), mannosidosis (3%), Wolman disease (2%), and other conditions (4%). The five-year OS has increased from 65% (95% confidence interval [CI], 52%-76%) in the historical cohort to 91% (95% CI, 81%-96%) in the current cohort (P<0.001). Moreover, the five-year ES, which was 64% (95 CI%, 56%-72%) for the entire cohort, has doubled from 41% (95% CI, 29%-53%) in the historical cohort to 85% (95% CI, 75%-92%) in the current cohort (P<0.001). The proportion of patients with graft failure has decreased from 37% in the historical cohort to 8% in the current cohort (P<0.001). In patients who received a second transplant, 13 out of 20 patients (65%) in the historical cohort and all four in the current cohort were alive and engrafted. Of 82 survivors followed-up at Manchester, 80% and 20% had full and mixed chimerism, respectively. Although this study was restricted to a single center, our findings show that HCT is an increasingly safe procedure and provides long-lasting endogenous enzyme replacement therapy for children with IMDs in the modern era of HCT.
Introduction
Inherited metabolic diseases (IMDs) are devastating multi-systemic disorders. They are associated with substantial morbidity, resulting in reduced quality of life and premature death in the severe phenotypes. Historically, the treatment of IMDs relied on symptomatic supportive care and palliative therapy, until a child with Hurler syndrome received the first successful hematopoietic cell transplantation (HCT) in 1980. The benefits of HCT mainly result from the endogenous production of a missing enzyme by circulating donor-derived leukocytes and the engraftment of donor-derived hematopoietic cells in the brain. The rationale for HCT is based on the principle of cross-correction. Through endocytosis, an enzyme secreted by donor cells is taken up by adjacent recipient cells, which prevents the accumulation of toxic metabolites and improves organ functions.
Although HCT can provide a long-lasting therapy for children with IMDs, its efficacy has been limited by high rates of graft failure, and transplant-related morbidity and mortality. Early studies reported a 5-year overall survival (OS) ranging from 50% to 80% and an engrafted survival (ES) ranging from 30% to 80%1. Since 1985, the Royal Manchester Children?s Hospital in the United Kingdom has devoted considerable effort to optimize the transplant care for children with IMDs. Here, we report and compare the OS, ES and transplant-related mortality (TRM) of two cohorts of children with IMDs (historical and current) who were treated in our metabolic transplant center.
Patients and Methods
From 1985 and 2016, 137 children with IMDs received their first HCT at the Royal Manchester Children?s Hospital. The clinical and laboratory data were retrieved from transplantation databases, patient medical files, and laboratory records. Donor chimerism data at the last evaluation were available for 82 patients who were alive and engrafted following first and second transplants. For all the patients, written informed consent was obtained from the parents or legal guardians.
In this study, the primary endpoints were OS (survival from the first HCT to the last evaluation or death), ES (probability of being alive with at least 20% donor-cell engraftment), and TRM (any death due to complications following the HCT). We also examined the diseaserelated mortality (DRM), which was defined as any death due to complications of the underlying primary condition. Other outcomes of interest included neutrophil recovery (first of three consecutive days with a neutrophil count ?0.5×109/L), platelet recovery (first day with a platelet count ?20×109/L followed by seven days without transfusion), transplant-related complications (venoocclusive disease, graft versus host disease [GvHD], and graft failure), and latest graft function (donor chimerism). The graft failures were categorized into four groups: primary aplasia (no neutrophil engraftment by day 42), primary autologous reconstitution (neutrophil engrafted by day 42 with less than 20% donor-derived hematopoiesis), secondary aplasia (sustained cytopenia following neutrophil engraftment with full donor-derived hematopoiesis), and secondary autologous reconstitution (falling donor chimerism [less than 20%] following neutrophil engraftment and adequate donor-derived hematopoiesis). Historical cohort comprised patients transplanted before 2007 while current cohort referred to patients transplanted from 2007 onwards.
The quantitative variables were described using the median, and 25th and 75th percentile (q25 and q75, respectively), while the categorical variables were reported with counts and percentages. The Wilcoxon rank-sum and Chisquare tests were used to analyze the continuous variables and compare the categorical variables, respectively. The probabilities of OS and ES were calculated using the Kaplan-Meier estimate. The two-sided log-rank test was used to compare the OS and ES between the historical and current cohorts. The cumulative incidence function (CIF) was used to estimate the probability of TRM, considering the DRM as the competing risk. Gray?s test was used to compare the TRM between the historical and current cohorts. All the reported p-values are two-sided with a level of significance set at 0.05. Statistical analyses were performed using STATA 14.2.
Results
Children with a primary diagnosis of mucopolysaccharidosis (MPS, 81%), X-linked adrenoleukodystrophy (X-ALD, 6%), metachromatic leukodystrophy (MLD, 4%), mannosidosis (3%), Wolman disease (WD, 2%), or other conditions (4%) were included the study. The characteristics of the patients and transplants are detailed in Table 1. We first examined the median time from diagnosis to transplant in children with MPS and found a reduction from 7.4 months in the historical cohort to 3.9 months in the current cohort (P<0.001). Moreover, we found that the affected children were transplanted at a younger age in the current cohort compared to the historical cohort (P=0.03). For surviving patients, the median follow-up time was 81 months (q25=6.3 months and q75=152.0 months). The data for hematopoietic recovery and early transplant-related complications are listed in Table 1. The median time to neutrophil recovery was 16 days and the median time to platelet recovery was 25 days. Our data also showed that in the current cohort, the incidence of grade III-IV acute GvHD and chronic GvHD were 6% and 2%, respectively.
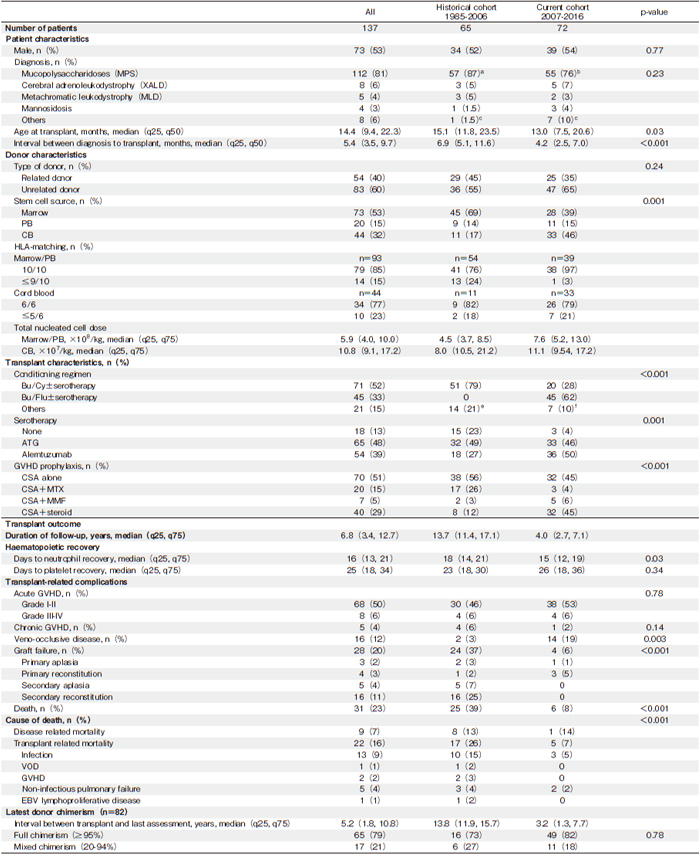
While the five-year estimated OS for the entire cohort was 79% (95% confidence interval [CI], 71%-85%), detailed analyses revealed that the five-year OS increased from 65% (95% CI, 52%-76%) for the historical cohort to 91% (95% CI, 81%-96%) for the current cohort (P <0.001) (Figure 1A). Moreover, for the transplanted children in the current cohort, subgroup analyses by primary diagnosis have shown that the five-year OS was 100% for X-ALD (n=5), 98% (95% CI, 83%-98%) for MPS (n=55), and 75% (95% CI, 42%-92%) for the other IMDs (mannosidosis, n=3; MLD, n=2; WD, n= 2; aspartylglucosaminuria, n=1; fucosidosis, n=1; glycogen storage disease 1B, n=1; Niemann-Pick disease C2, n=1; and Tay-Sachs disease, n=1).
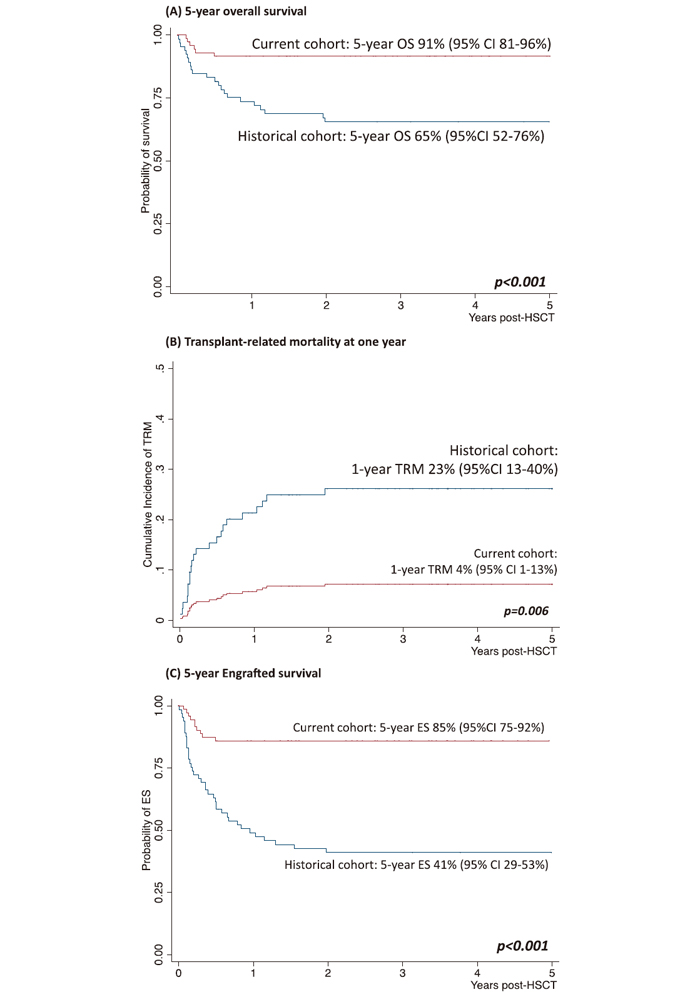
Using the CIF, we also found that the one year TRM probability for the entire cohort was 13% (95% CI, 4%-14%), while detailed analyses show that the one year TRM probability decreased from 23% (95% CI, 13%- 40%) for the historical cohort to 4% (95% CI, 1%-13%) for the current cohort (P=0.006) (Figure 1B). In the entire cohort, two of the eight transplanted patients with X-ALD died of advanced disease, while the other six were alive and engrafted. Moreover, deaths due to transplant- related complications have decreased significantly in the current cohort (n=6, 8%) than in the historical cohort (n=25, 39%) (P<0.001).
On analyzing the entire cohort, we found that the fiveyear ES was 64% (95 CI%, 56%-72%). However, when we compared the two cohorts, we found that the ES had doubled from 41% (95% CI, 29%-53%) in the historical cohort to 85% (95% CI, 75%-92%) in the current cohort (P<0.001) (Figure 1C). The frequency of graft failure for the entire cohort was 20%, significantly decreasing from 37% in the historical cohort to 8% in the current cohort (P<0.001) (Table 1). In the entire cohort, 28 patients had graft failure, while 24 of these patients received a second HCT, two died of disease-related complications, and the other two were lost to follow-up. At the time of the study, 17 of the patients who received a second transplant were alive and engrafted, 13 out of 20 (65%) for the historical cohort and all four (100%) for the current cohort. Among the seven patients who died after a second transplant, five died of transplant-related complications (infection, n=2; GvHD, n=2; EBV lymphoproliferative disease, n=1) and two died of disease progression (MLD, n=1; MPS type III, n=1). For the entire cohort of patients who received a second HCT, the estimated 5-year ES was 79% (95% CI, 56%-91%), increasing from 65% (95% CI, 35%-83%) for the historical cohort to 100% for the current cohort (P=0.29).
Finally, we examined the donor chimerism data at the last evaluation. Out of 82 patients who were alive and engrafted following a first or second transplant (Table 1), 65 (79%) had full-donor chimerism, while the remaining 17 (21%) had mixed-donor chimerism.
Discussion
A successful HCT provides a long-lasting diseasemodifying treatment and improves the long-term survival of children with IMDs. The donor-derived leukocyte engraftment provides a stable source of enzyme for the in–situ cross-correction of neighboring tissues, including neurons, and has an immunomodulatory effect if there is a significant inflammatory component to the disease. Here, we presented a detailed analysis of the clinical outcomes of children with IMDs, all treated in a single metabolic center over the last three decades. Importantly, our center has treated twice as many children in the last decade (n=72) than in the previous two decades (n=65). This doubling of the rate of HCT for the treatment of IMDs can be attributed to 1) an increased global awareness of the potential of HCT for IMDs, 2) improvements in diagnostic methods and family and newborn screening, and 3) a progressive centralization of referrals, resulting in an increase in the number of patients treated in our center. The comparison of our historical and current cohorts illustrates the treatment progress and improvements in overall and engrafted survivals of the children with IMDs treated in our center. The factors that contributed to this improvement included greater availability of well-matched unrelated donors in worldwide registries, better supportive care, more effective anti-microbial therapy, and the implementation of international HCT guideline for IMDs (including busulfan pharmacokinetic monitoring).
Following the international collaborative efforts to examine transplant and disease outcomes, there has been a change in the indication of HCT and a shift in the donor cell source. It is essential to distinguish the transplant outcomes from the disease outcomes to optimize the transplant care for children with IMDs. Besides achieving superior transplant survival rates, the studies carried over the last three decades have also provided us with a learning platform to understand the therapeutic efficacy of HCT in IMDs. To date, it has been shown that some IMDs, including Hurler syndrome, X-ALD, MLD, and globoid cell leukodystrophy, are amenable to HCT. Conversely, other IMDs, including MPS type III and GM gangliosidosis, do not respond to HCT, and the diseases continue to progress in patients transplanted before the symptomatic phase, with patterns similar to those observed in untransplanted patients2. The hypotheses proposed to explain treatment failure in HCT are as follows: 1) an insufficient production of enzyme by the donorderived microglia in the brain and 2) a neurological deterioration occurring before the progressive cell replacement process. Some of these limitations may be addressed by ex vivo gene therapy in hematopoietic stem cells (HSCs), using a lentiviral vector strategy. Indeed, we have previously demonstrated the efficacy of lentiviral vector-modified HSCs in mouse models of MPS type IIIA and IIIB. Moreover, these findings have been translated into a clinical trial for MPS type IIIA3-5. This strategy has now entered late-phase clinical trials in children with MLD, demonstrating the safety and therapeutic benefits of ex vivo gene therapy in HSCs6,7.
The significant improvements in transplant outcomes have provided some insights into the exploration of cellular therapy as a potentially curative treatment for children with IMDs. Although the cohort analyzed here was restricted to patients treated in a single center, our findings have confirmed that HCT, when performed in an experienced metabolic transplant center, is an increasingly safe procedure and provides long-lasting therapy for children with IMDs. These results will contribute to a reevaluation of the potential role of transplant in such centers, particularly for the treatment of diseases, in which the perceived risk that is often based on survival data from a previous era, prevents the use of transplant as a therapy. It will also be interesting to consider transplant as an alternative therapy for diseases where enzyme replacement therapy is currently the standard treatment, such as attenuated MPS type I and MPS type II. Collaborations between multidisciplinary teams and international metabolic transplant centers will be critical in promoting the progress of cellular therapies.
Acknowledgments
We would like to thank all the patients, their families, and the treating multidisciplinary team.
Authors’ contribution
S. H. L. conceptualized and designed the study, performed the statistical analysis, and wrote the first draft of the manuscript. M. C, H. J. C., K. L. T., J. M., W. O., H. L., A. L., and T. K. contributed to the data collection. R. F. W., S. A. J., and P. H. critically reviewed the final manuscript. All authors (included A. W., D. B. and S. R.) edited and approved the manuscript.
Conflict of Interests
The authors declare no conflict of interest. Disclosure forms provided by the authors are available here.
References
1. S. H. Lum SAJ, A. Ghosh, B. W. Bigger, R. F. Wynn. Hematopoietic stem cell transplant for the mucopolysaccharidoses. Expert Opinion on Orphan Drugs. 2016; 4: 379-393.
2. Boelens JJ, Orchard PJ and Wynn RF. Transplantation in inborn errors of metabolism: current considerations and future perspectives. Br J Haematol. 2014; 167: 293-303.
3. Langford-Smith A, Wilkinson FL, Langford-Smith KJ, Holley RJ, Sergijenko A, Howe SJ, et al. Hematopoietic stem cell and gene therapy corrects primary neuropathology and behavior in mucopolysaccharidosis IIIA mice. Mol Ther. 2012; 20: 1610-21.
4. Sergijenko A, Langford-Smith A, Liao AY, Pickford CE, McDermott J, Nowinski G, Langford-Smith KJ, et al.. Myeloid/Microglial driven autologous hematopoietic stem cell gene therapy corrects a neuronopathic lysosomal disease. Mol Ther. 2013; 21: 1938-49.
5. Brian Bigger SE, Daniel Fil, Claire O?leary, John McDermott, N. Senthivel, Alexander Langford-Smith, et al. Neurological correction of mucopolysaccharidosis type IIIB mice by haematopoietic stem cell gene therapy. Mol Genet Metab. 2017; 120: S28.
6. Sessa M, Lorioli L, Fumagalli F, Acquati S, Redaelli D, Baldoli C, et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet. 2016; 388: 476-87.
7. Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013; 341: 1233158.
Search
News


