Volume 2 (2019) Issue 1 No.2 Pages 5-8
Abstract
The aggressive variant of large granular lymphocyte (LGL) leukemia is very rare and the prognosis of this disease is poor. A 47-year-old woman with progressive pancytopenia and severe liver damage visited our institute. Upon hospitalization, about 30% LGL was detected in her peripheral blood and bone marrow samples. Flow cytometry was conducted to analyze lymphocytes in the bone marrow, which revealed the presence of CD3 and T-cell receptor (TCR) α/β and absence of CD4, CD8, CD16, CD56, CD22, CD79a, and terminal deoxynucleotidyl transferase (TdT). Southern blotting was performed, which revealed the presence of rearrangement of TCR-Cβ1 and Jγ. We made a diagnosis of the aggressive variant of T-LGL leukemia, and performed myeloablative allogeneic peripheral stem cell transplantation (allo-HSCT) from an HLA-matched sibling for primary refractory disease of CHOP and hyper CVAD therapy. She is alive in remission with donor-derived T-LGL lymphocytosis in peripheral blood for 7 years after allo-HSCT. Overall, Allo-HSCT could be active against the aggressive variant of LGL leukemia and induce graft-versus-leukemia effect.
Introduction
Large granular lymphocyte (LGL) leukemia is a rare indolent hematological disorder derived from CD3+ cytotoxic T cells or CD3- NK cells. Although standard treatment has not been established, immunosuppressive drugs, such as cyclophosphamide, methotrexate, or cyclosporine, are administered for indolent LGL leukemia. However, a type of this disease is highly aggressive and accompanied by B symptoms, lymphocytosis, hepatosplenomegaly, lymphadenopathy, and severe cytopenia, and is known to be the aggressive variant1,2. Treatment for the aggressive variant of LGL leukemia, which includes allogeneic hematological stem cell transplantation (allo-HSCT), is similar to that for acute lymphoblastic leukemia1. In this study, we report a rare case of allo-HSCT for a patient with primary refractory aggressive variant of T-LGL leukemia, resulting in long-term survival.
Case presentation
A 47-year-old woman visited our institute because pancytopenia and severe liver damage were detected in November 200X. A morphological examination of her peripheral blood (PB) and bone marrow revealed large proportions of abnormal lymphocytes, that is, 30% and 35%, respectively (Figure 1).
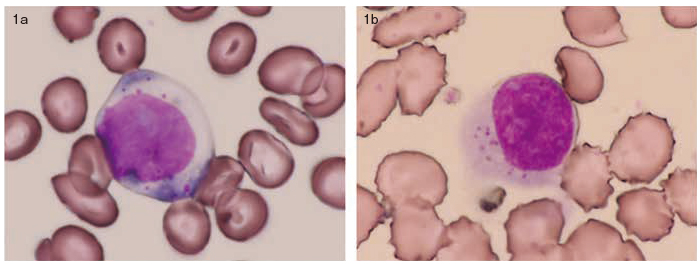
Hematological examination revealed a leukocyte count of 1100 cells/μL, including a neutrophil count of 260 cells/μL. The lymphocyte count was 820 cells/μL, including an LGL count of 420 cells/μL. Biochemical tests revealed levels of aspartate aminotransferase, alanine transferase, and lactate dehydrogenase to be 1010 IU/l, 666 IU/l, and 5330 IU/l, respectively.
Flow cytometry was conducted to analyze lymphocytes in the bone marrow, which revealed the presence of CD3 and T-cell receptor (TCR) α/β as well as absence of CD4, CD8, CD16, CD56, CD22, CD79a, and terminal deoxynucleotidyl transferase (TdT). Southern blotting was then performed, which revealed the presence of TCR-Cβ1 and Jγ rearrangements. A CT scan of the abdomen revealed that the spleen was 11 cm in size, and lymph nodes were not swollen. Pancytopenia and liver damage developed progressively within a week. Therefore, the patient was diagnosed with an aggressive variant of T-LGL leukemia.
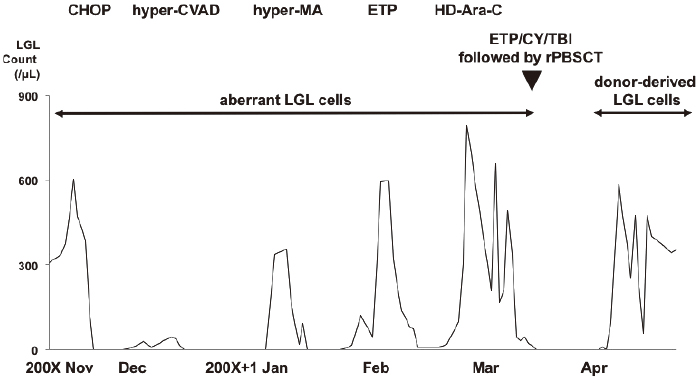
CHOP therapy was initiated, and LGLs were not detected in the PB sample and liver function improved. Since CHOP was a low intensive regimen for the aggressive variant of T-LGL leukemia, we changed the chemotherapy to a hyper-CVAD/MA regimen. However, after hyper-MA therapy, the LGL count increased to 600 cells/μL in the PB sample, and pancytopenia progressed. We then planned to conduct allo-HSCT for the primary refractory aggressive variant of T-LGL leukemia. Etoposide monotherapy (etoposide, 500 mg/m2, day 1-4) and Ara-C monotherapy (cytarabine, 1 g/body, day 1) were performed to reduce the tumor burden prior to the allo-HSCT.
We performed allo-HSCT from an HLA-matched male sibling. The conditioning regimen included total body irradiation (total 10 Gy) and administration of etoposide (15 mg/kg for two days) and cyclophosphamide (60 mg/kg, for two days). We infused 4.20×106/kg of CD34+ cells. Short-term administration of methotrexate and cyclosporine was used as prophylaxis for graft-versus-host disease (GVHD).
On day 14, neutrophil engraftment was observed; however, LGLs were detected in the PB sample simultaneously. On day 15, chimerism analysis of the PB sample revealed XY of count was 98.4% by fluorescence in situ hybridization. After engraftment, the LGL count changed from 240 cells/μL to 2300 cells/μL in the PB sample. However, their phenotype was different from that of the malignant LGLs; CD3, CD45RA, CD57, CD62, and TCR-αβ were detected using flow cytometry. Either CD4 or CD8 was present, and CD56 was absent. TCR-β rearrangement was not detected. We repeated the CD3 chimerism analysis of the PB sample, which revealed complete chimerism at all times. Thus, those LGLs had different characteristics from the native malignant LGLs, and were donor-derived LGLs without the clonality. The patient was discharged on day 50, and she discontinued cyclosporine on day 210. LGLs have been detected in the PB sample since then. She is alive with LGLs in the PB sample for 7 years after allo-HSCT. The clinical course, focusing on he count of LGLs, is shown in Figure 2.
Discussion
We reported that allo-HSCT was effective in a patient with a primary refractory aggressive variant of T-LGL leukemia. Donor-derived LGLs were detected, which induced graft versus leukemia (GVL) effects.
Marchand et al. (2016) reported the outcome of LGL leukemia patients as performed with HSCT using European Society for Blood and Marrow Transplantation (EBMT) registry3. The authors demonstrated that the 2-year progression-free survival and overall survival rates were 50% each in 10 patients treated with allo-HSCT; four patients died from severe infection and one of progressive disease. Furthermore, the authors suggested that autologous-HSCT and allo-HSCT were suitable for patients with chemo-sensitive disease and chemo-refractory disease, respectively.
The GVL effect is an immunological activity via donor cytotoxic T cells and NK cells after allogeneic HSCT. The GVL effect is induced by activation of donor T-cell and NK cell via mismatch of HLA and KIR4.
Several studies have demonstrated that LGLs, which are benign cytotoxic T-cells or NK cells, mediate a cytotoxic activity in malignant cells after allo-HSCT5,6. Gill et al. demonstrated that the frequency of T-LGL leukemia was 0.5% among patients treated with allo-HSCT7. Dhodapkar et al. (1994) proposed T-cell clonopathy of undetermined significance, which was shown as asymptomatic neoplastic T-LGL proliferation8. However, the diagnostic criteria of T-cell clonopathy of undetermined significance has not been defined. In addition, asymptomatic T-LGL lymphocytosis was reported in patients treated with renal and cardiac transplantation9,10. In this case, LGLs in the PB sample after allo-HSCT did not include malignant clones because the phenotype of the LGLs was different from those of the T-LGL cells at diagnosis, TCR rearrangement was not detected, and the results of chimerism analysis in CD3+T cells revealed complete chimerism at all times after allo-HSCT. We identified the LGLs after engraftment as benign T-cells, which contributed to the GVL effects.
In conclusion, we report a case with the aggressive variant of T-LGL leukemia that was managed by allo-HSCT. We suggest that allo-HSCT is necessary to improve the outcome in patients with the aggressive variant of T-LGL leukemia. LGLs in the PB sample after allo-HSCT were benign mature T cells, which might be a contributing factor in the long relapse-free survival as a GVL effect.
Acknowledgment
The authors thank Koji Sano for his assistance in the care of this patient,
Authors’ Contribution
KS, KN, and JM cared for the patient and provided clinical data and materials. KS, KN, JM, HM, MK and HY analyzed and interpreted routine diagnostic results. KS, KN, and SY wrote the manuscript. All authors read and approved the final manuscript.
Financial Support
None
Conflict of Interest
The authors have no conflict of interests to declare.
References
1. Lamy T, Loughran TP. Large granular lymphocyte leukemia. Cancer Control. 1998; 5: 25-33.
2. Ruskova A, Thula R, Chan G. Aggressive natural killer-cell leukemia: report of five cases and review of the literature. Leuk Lymphoma. 2004; 45: 2427-38.
3. Marchand T, Lamy T, Finel H, Arcese W, Choquet S, Finke J, et al. Hematopoietic stem cell transplantation for T-cell large granular lymphocyte leukemia: a retrospective study of the European Society for Blood and Marrow Transplantation. Leukemia. 2016; 30: 1201-4.
4. Bleakley M, Riddell SR. Molecules and mechanisms of the graft-versus-leukemia effect. Nat Rev Cancer. 2004; 4: 371-80.
5. Mohty M, Faucher C, Vey N, Chabannon C, Sainty D, Arnoulet C, et al. Features of large granular lymphocytes (LGL) expansion following allogeneic stem cell transplantation: a long-term analysis. Leukemia. 2002; 16: 2129-33.
6. Au WY, Lam CC, Lie AK, Pang A, Kwong YL. T-cell large granular lymphocytes leukemia of donor origin after allogeneic bone marrow transplantation. Am J Clin Pathol. 2003; 120: 626-30.
7. Gill H, Ip AH, Leung R, So JC, Pang AW, Tse E. Indolent T-cell large granular lymphocyte leukemia after haematopoietic SCT: a clinicopathologic and molecular analysis. Bone Marrow Transplant. 2012; 47: 952-6.
8. Dhodapkar MV, Li CY, Lust JA, Tefferi A, Phyliky RL. Clinical spectrum of clonal proliferations of T-large granular lymphocytes: a T-cell clonopathy of undetermined significance? Blood. 1994; 84: 1620-7.
9. Gentile TC, Hadlock KG, Uner AH, Delal B, Squiers E, Crowley S, et al. Large granular lymphocyte leukemia occurring after renal transplantation. Br J Haematol. 1998; 101: 507-12.
10. Sabnani I, Zucker MJ, Tsang P, Palekar S. Clonal T-large granular lymphocyte proliferation in solid organ transplantation recipients. Transplant Proc. 2006; 38: 3437-40.
Search
News


