Volume 2 (2019) Issue 1 No.1 Pages 1-4
Abstract
Neurological complications after hematopoietic stem cell transplantation (HSCT) are frequently life-threatening, and their clinical management can be highly challenging. In the case of central nervous system lesions post-HSCT, a definitive diagnosis is often difficult to reach because many different causative and contributing conditions may be present, including bacterial, fungal, or viral infections; original disease relapse; and post-transplant lymphoproliferative disorder (PTLD). Here, we report a case of a 32-year-old male patient with Philadelphia chromosomepositive acute lymphoid leukemia who underwent three HSCTs and was then diagnosed with primary central nervous system (PCNS) PTLD by brain biopsy. The third HSCT was a haplo-identical peripheral blood stem cell transplantation from his mother, with post-transplant high-dose cyclophosphamide and tacrolimus used as graft-versus-host disease prophylaxis. Four months after the HSCT, multiple small ring lesions were detected in the parabasal ganglia of the patient’s brain during magnetic resonance imaging. A lesion biopsy indicated Epstein-Barr virus (EBV)-positive, diffuse large B-cell lymphoma. Because the patient had no evidence of systemic lymphadenopathy, we diagnosed him with PCNS-PTLD. There was no EBV DNA in this patient’s cerebrospinal fluid. The diagnosis of PCNS-PTLD by EBV DNA polymerase chain reaction is difficult and highlights the importance of a brain biopsy to diagnose PCNS-PTLD, especially in cases showing no EBV DNA in the cerebrospinal fluid. Although a rare condition, it is essential to locate and analyze cases of PCNS-PTLD after HSCT to establish the optimal strategy for treatment or prophylaxis.
Introduction
Post-hematopoietic stem cell transplantation (HSCT) lesions in the central nervous system (CNS) are generally difficult to diagnose definitively because of the many different conditions that could be present, such as bacterial, fungal, or viral infections; original disease relapse; and post-transplant lymphoproliferative disorder (PTLD). Here, we report multiple CNS lesions due to PTLD in a patient with Philadelphia chromosome-positive acute lymphoid leukemia (Ph+ ALL) after haplo-identical HSCT using post-transplant high-dose cyclophosphamide (PT-Cy).
The estimated incidence of PTLD in the transplantation population ranges from 1.2 to 11.2%1. PTLD typically develops in the first 6 months post-transplantation. Extra-nodal involvement is common in PTLD, although the CNS is an uncommon location, particularly in isolation. Primary CNS (PCNS) PTLD is clinically aggressive lymphoma of donor origin, with monomorphic histology in 83% and Epstein-Barr virus (EBV) +tumors in 94% cases2. The prognosis of patients with PCNS-PTLD is poor; furthermore, to date, there is no standard recommended therapy.
Case presentation
A 32-year-old man with Ph+ ALL underwent three HSCTs. The first was an unrelated bone marrow transplantation (BMT) from a human leucocyte antigen (HLA) 5/6-matched donor in his first complete remission (CR). The patient’s leukemia relapsed 1 year after HSCT. He received the second HSCT using HLA 4/6-matched cord blood in his hematological non-CR. The patient again experienced relapse, four months after the second transplantation. We detected a T315I mutation in the relapsed cells and initiated ponatinib administration. Because of the disease progression, the patient received a haplo-identical peripheral blood stem cell transplantation from his mother (HLA 3/6 matched) as the third HSCT. The conditioning regimen was busulfan and melphalan without CNS prophylaxis. The patient received PT-Cy (50 mg/kg) and tacrolimus as GVHD prophylaxis. He exhibited skin stage-2, gut stage-3, grade-? acute GVHD (aGVHD), which was treated with tacrolimus+prednisolone 1 mg/kg.
Four months after the third HSCT, the patient had a convulsion. Magnetic resonance imaging (MRI) revealed multiple small CNS ring lesions (Figure 1). After emergency admission, the physical examination showed a Glasgow Coma Scale score of 4-4-6, drowsy consciousness, no neck stiffness and skin sclerosis due to chronic GVHD. The patient’s blood examination yielded the following information: white blood cell count, 9900/uL (segs: 49%, stabs: 1.0%, lymphocytes: 38%, monocytes: 7%, eosinophils: 4%); hemoglobin, 8.3 g/dL; platelets, 4.3×104/uL; aspartate aminotransferase, 89 U/L; alanine aminotransferase, 50 U/L; lactate dehydrogenase, 299 U/L; alkaline phosphatase, 908 U/L; total bilirubin, 0.6 mg/dL; Renal function tests were normal. A cerebrospinal fluid (CSF) examination indicated the following; total protein, 81 mEq/L; glucose, 30 mg/dL; cell count, 1; adenosine deaminase, 3.7 IU/L; negative tuberculosispolymerase chain reaction (PCR); negative toxoplasma-PCR; negative EBV-PCR; negative minor bcr/abl; and negative culture. No malignant cells were detected by cytology. We could not perform biopsy at that time because these lesions were located in the deep part of the CNS.
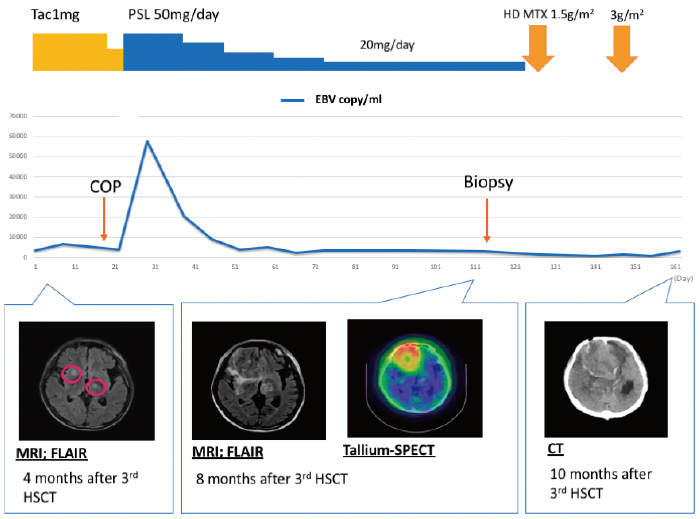
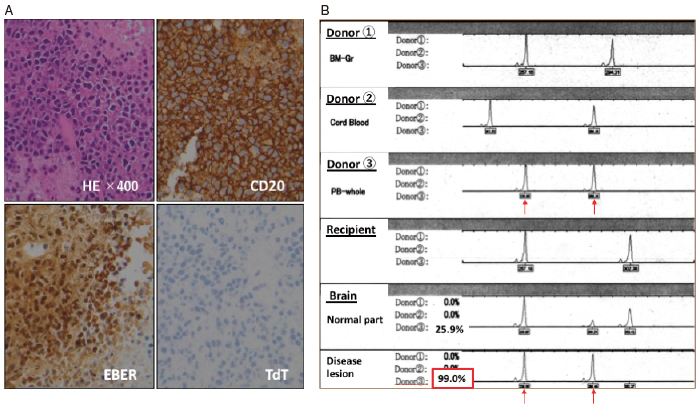
Based on MRI findings, we suspected that the lesions were nocardiosis or toxoplasmosis; thus, we prescribed sulfamethoxazole/trimethoprim. However, the CNS lesions grew to a maximum diameter of 56 mm in the patient’s right frontal lobe by 8 months after the third HSCT. A biopsy from the region revealed a diffuse pattern of hyperchromatic, large lymphoid cells. Immunohistochemical staining revealed the lymphoid cells to be positive for CD20,79a, BCL-2, MUM1, OCT-2, Epstein-Barr encoding region RNA in situ hybridization, LMP-1, and EBNA2, and negative for CD3, CD5, CD10, and bcrabl. Thus, this lesion was diagnosed as EBV-positive diffuse large B-cell lymphoma (Figure 2-A); the overall features were consistent with PTLD. We then reduced the immunosuppressive agents. The patient received highdose methotrexate (1.5 g/m2 and 3 g/m2) therapy, which was only temporarily effective. He subsequently died from PCNS-PTLD. The autopsy revealed no evidence of systemic PTLD. Short tandem repeat analysis confirmed that the PTLD cells originated from the mother as donor 3 (Figure 2-B).
Discussion
There are two published case series of PCNS-PTLD after solid organ transplantation2,3; however, there are no case series of this disease after HSCT. PCNS PTLD presents with nonspecific clinical symptoms; the typical imaging findings include multiple bilateral hemispheric lesions with involvement of the subcortical white matter and basal ganglia. Radiographically, all lesions are generally enhanced either homogenously or in a ring-enhancing pattern3.
Monitoring of EBV DNA in whole blood showed no parallels with the clinical symptoms in our case (Figure 1), and there was no EBV DNA in this patient’s CSF. Some case reports showed positive results for EBV DNA in the CSF of patients with CNS-PTLD4,5, and most patients test positive for EBV DNA in peripheral blood. In contrast, some authors have reported EBV DNA-negative blood and CSF in patients with CNS PTLD6, suggesting that the diagnosis of PCNS-PTLD by EBV DNA PCR is difficult. These reports highlight the importance of a brain biopsy to diagnose PCNS-PTLD, especially in cases showing no EBV DNA in the CSF.
To date, no standard therapy for PCNS-PTLD has been recommended. Possible therapeutic options include (?) chemotherapy±rituximab in line with primary CNS lymphoma protocols, (?) monotherapy with rituximab, (?) T-cell therapy with EBV-specific cytotoxic T lymphocytes, and (?) radiotherapy1.
The risk of PTLD is increased with unrelated and HLA-mismatched (>2) related donor bone marrow, use of anti-thymocyte globulin or anti-CD3 monoclonal antibody for prophylaxis or treatment of acute GVHD7, cord blood transplantation, T-cell depletion, EBV serology donor-recipient mismatches, splenectomy, and second HSCT1. To our knowledge, this is the first case report of PCNS-PTLD after haplo-identical HSCT with PT-Cy. Interestingly, there is a report about absence of PTLD after allogeneic bone marrow transplantation using PT-Cy8. Our report shows that the possibility of PTLD should be considered even after such therapy. In the future, it will be essential to locate and analyze cases of PCNS-PTLD after HSCT to establish the best strategy for treatment or prophylaxis.
Authors’ Contributions
M. T. wrote the manuscript. J. A., S. S., Y. A., S. M., H. K., H. M., D. O., K. M., and S. K. collected the data. Y. K., H. K., and N. N. analyzed the pathology. M. O., K. A., K. H., Y. O., and H. K. revised the manuscript.
Conflict of Interest
The authors declare no conflict of interest.
References
1. Styczynski J, van der Velden W, Fox CP, Engelhard D, de la Camara R, Cordonnier C, et al. Management of Epstein-Barr Virus infections and post-transplant lymphoproliferative disorders in patients after allogeneic hematopoietic stem cell transplantation: Sixth European Conference on Infections in Leukemia (ECIL-6) guidelines. Haematologica. 2016; 101: 803-11.
2. Evens AM, Choquet S, Kroll-Desrosiers AR, Jagadeesh D, Smith SM, Morschhauser F, et al. Primary CNS posttransplant lymphoproliferative disease (PTLD): an international report of 84 cases in the modern era. Am J Transplant. 2013; 13: 1512-22.
3. Cavaliere R, Petroni G, Lopes MB, Schiff D. International Primary Central Nervous System Lymphoma Collaborative Group. Primary central nervous system post-transplantation lymphoproliferative disorder: an International Primary Central Nervous System Lymphoma Collaborative Group Report. Cancer. 2010; 116: 863-70.
4. Hoshino Y, Kimura H, Tanaka N, Tsuge I, Kudo K, Horibe K, et al. Prospective monitoring of the Epstein-Barr virus DNA by a real-time quantitative polymerase chain reaction after allogenic stem cell transplantation. Br J Haematol. 2001; 115: 105-11.
5. Yaginuma T, Yamamoto H, Mitome J, Tanno Y, Yamamoto I, Kobayashi A, et al. Successful treatment of monomorphic primary central nervous system post-transplantation lymphoproliferative disorder 5 years after kidney transplantation. Transpl Infect Dis. 2012; 14: E102-6.
6. Hamadani M, Martin LK, Benson DM, Copelan EA, Devine SM, Hofmeister CC. Central nervous system post-transplant lymphoproliferative disorder despite negative serum and spinal fluid Epstein-Barr virus DNA PCR. Bone Marrow Transplant. 2007; 39: 249-51.
7. Curtis RE, Travis LB, Rowlings PA, Socie G, Kingma DW, Banks PM, et al. Risk of lymphoproliferative disorders after bone marrow transplantation: a multi-institutional study. Blood. 1999; 94: 2208-16.
8. Kanakry JA, Kasamon YL, Bolanos-Meade J, Borrello IM, Brodsky RA, Fuchs EJ, et al. Absence of post-transplantation lymphoproliferative disorder after allogeneic blood or marrow transplantation using post-transplantation cyclophosphamide as graft-versus-host disease prophylaxis. Biol Blood Marrow Transplant. 2013; 19: 1514-17.
Search
News


