Volume 3 (2020) Issue 3 No.3 Pages 48-58
Abstract
We report a single-center experience of allogeneic hematopoietic stem cell transplant for patients with severe aplastic anemia over 13 years of age. Fifty-eight patients were included, and in vivo T-cell depletion was used in all cases. Fifty-one (88%) received alemtuzumab and 7 (12%) were given rabbit anti-thymocyte globulin. The median follow-up period was 6 years (range: 0-13.5). Data was collected retrospectively and updated in April 2019. The median age was 31 years (range: 18-67). Forty (69%) recipients received grafts from related donors and 18 (31%) received them from unrelated donors. Peripheral blood grafts were infused in 12 (20.7%) patients. Five-year overall survival (OS) was 80.7%. Five-year graft-versus-host disease (GVHD) /rejection-free survival was 56%. Eight (13.8%) patients experienced graft failure. The cumulative incidence of grade ?-? acute GVHD at day 100 was 14% and that of chronic GVHD at 1 year was 7%. The selection of unrelated donors and the use of peripheral blood grafts were not significant risk factors for clinically relevant GVHD or for lower OS. Recipients older than 40 years showed significantly worse OS, as observed from the results of univariate analysis. T-cell depletion in severe aplastic anemia shows low rates of GVHD and high OS, but older patients remain a group with higher risk of mortality. Long-term complications were mainly autoimmune in character.
Introduction
One therapeutic option for patients with severe aplastic anemia (SAA) is allogeneic hematopoietic stem cell transplantation (alloHSCT), either upfront or after failure, to respond to immunosuppressive therapy (IST) 1. The choice of the timing for alloHSCT depends on the recipient?s age, comorbidities, and the availability of a matched related donor1,2 There is no strict upper age limit for alloHSCT. However, decisions are taken on an individual basis, as the outcome of patients over 40 years is inferior compared with that of younger patients with SAA3-8.
The goal of alloHSCT in SAA is to achieve sustained engraftment with minimal transplant-related toxicities and graft-versus-host disease (GVHD). There is significant variation between centers, according to the choice of conditioning regimens8-11. The most frequent preparative regimen is cyclophosphamide (Cy), alone or in combination with anti-thymocyte globulin (ATG) or alemtuzumab. For GVHD prophylaxis, methotrexate (MTX) is frequently combined with cyclosporine (CsA) 12-19. The increased use of T-cell depletion as part of the conditioning regimen has contributed to the reduction of clinically relevant GVHD. However, despite the refinement of GVHD prophylaxis, around 15-25% of patients still develop GVHD, resulting in impairment of quality of life, morbidity, and mortality20-23.
Here, we report the outcomes and experiences of using in vivo T-cell depletion as part of the conditioning regimen for SAA, in a single, large center. This study includes 41 patients with SAA previously reported, with an updated, long-term follow-up14.
Patients and Methods
Patient Selection
Between January 2005 and April 2019, 58 consecutive adult patients with SAA underwent alloHSCT at the Princess Margaret Cancer Center, Ontario. Data was collected through retrospective chart review. The last follow-up was in April 2019.
According to international guidelines for management of SAA1-4, upfront alloHSCT is indicated for patients younger than 40 years who have a 10/10 matched sibling donor (MSD). For older recipients or those without a 10/ 10 MSD available, alloHSCT using a 10/10 matched, unrelated donor (MUD), or an alternative donor such as a 9/10 MUD may be considered, after failure to respond to or relapse after ATG-containing immunosuppressive therapy (IST). This study was conducted in accordance with the Declaration of Helsinki, and reviewed and approved by the Health Network-Research Ethics Board and Cancer Registry Data Access Committee of the University of Toronto.
Donor Selection and Graft Source
Forty patients (69%) received grafts from a 10/10 MSD. Seventeen patients (29%) received 10/10 MUD grafts, and a 9/10 matched-unrelated donor was used for a single case (2%). No haploidentical donors were selected during this period of time. A bone marrow (BM) source was the preferred option, upfront. However, peripheral blood grafts were accepted, according to each donor?s choice.
Conditioning Regimen, Post-transplant Follow-up, and Supportive Care
Between January 2005 and December 2015, 8 patients (14%) received high-dose Cy (50 mg/kg ? daily from day -5 to -2) and alemtuzumab (Cam) 50 mg (10 mg, 20 mg and 20 mg daily on days -8, -7, and -6, respectively) (Cy-Cam). Forty recipients (69%) received fludarabine (Flu) (30 mg/m2 ? daily from day -5 to -2), low-dose Cy (10 mg/kg ? daily from day -5 to -2) and Cam 60 mg (30 mg daily on days -7 and -6) (Flu-Cy-Cam). Busulfan (Bu) replaced Cy in the Flu- Cy-Cam regimen in 3 patients (5%). Low dose total body irradiation (TBI) (200 cGy) was given on day -1 to 18 patients (31%) who received MUD grafts.
From January 2016, alemtuzumab was replaced by rabbit-ATG (Thymoglobulin; Genzyme-Sanofi, Lyon, France) in combination with Flu and Cy as part of the standard conditioning platform in SAA (Flu-Cy-ATG). This strategy continues to be the standard regimen for these patients at the Princess Margaret Cancer Center. ATG is administered in doses of 0.5 mg/kg on day -3, 2 mg/kg on day -2, and 2 mg/kg on day -1 (total dose 4.5 mg/kg). Six patients (10%) received this conditioning regimen. Bu replaced Cy in the Flu-Cy-ATG regimen in a single case (2%).
In all cases, T-cell-replete grafts were infused on day 0. All patients received granulocyte-colony stimulating factor (G-CSF) 300 mcg subcutaneously, daily from day +7 until the neutrophil count was>1.5×109/L. Prophylactic antimicrobial therapy included posaconazole 300 mg daily, acyclovir 400 mg twice daily, ciprofloxacin 500 mg twice daily, and inhaled pentamidine 300 mg monthly which was replaced by oral trimethoprim-sulfamethoxazole (one double-strength tablet three times a week), when blood counts were stable. Post engraftment, cytomegalovirus (CMV) titer was monitored twice per week and Epstein-Barr virus (EBV) weekly.
Graft versus host disease prophylaxis
From 2005 to December 2015, 38 recipients (66%) received alemtuzumab, CsA and MTX for GVHD prophylaxis. CsA was started at a dose of 2.5 mg/kg/day ?, every 12 hours, from day -1, and adjusted to achieve a therapeutic level between 200-400 mcg/L. MTX was given at a dose of 15 mg/m2 ? on day +1 and 10 mg/ m2 on day +3 and +6. Eleven patients (19%) received tacrolimus (titrated to therapeutic levels of 10 to 20μg/L) instead of CsA, due to a history of gum hypertrophy secondary to CsA (10 patients) or intolerance (1 patient). Two recipients (3%) received mycophenolate mofetil (MMF), primarily due to end-stage kidney disease at the time of alloHSCT, and secondary to prior history of posterior reversible encephalopathy syndrome (PRES).
Between September 2013 and May 2018, 6 patients (8%) received ATG, CsA and MTX for GVHD prophylaxis. PTCy (50 mg/kg ? on day +3 and +4) was given, instead of MTX, to 1 patien (t 2%), who had a prior diagnosis of Hodgkin lymphoma (HL), and was subsequently diagnosed with SAA. Although the HL was in complete remission, and the alloHSCT was done secondary to the SAA, PTCy was given in combination with ATG and CsA because it was the standard GVHD prophylaxis used for patients diagnosed with hematological malignancies undergoing reduced intensity conditioning (RIC) for alloHSCT at our center. In addition, this prophylaxis was selected according to the hypothesis that PTCy would increase the cytotoxic effect of the reduced intensity regimen. Immunosuppression tapering was initiated at 6 months after alloHSCT, in all cases, unless mixed chimerism or active GVHD was present.
Disease monitoring
GVHD-Free/Rejection-free survival (GRFS) was calculated in months from the date of alloHSCT to the date of the event or last follow-up whichever came first. Grades ?-? acute GVHD, any grade of chronic GVHD, graft failure, and death were recorded as events, as SAA is a non-malignant disorder25,26. Long-term complications were studied through retrospective chart review. They were stratified between non-malignant complications and secondary neoplasia. Complications, management and outcome are detailed in Results Section. The definitions of engraftment, graft failure, and overall survival are to be found in the supplementary material.
Statistical Methods
Baseline characteristics were reported using descriptive statistics. OS and GRFS rates were calculated using the Kaplan-Meier product-limit method and the impact of variables was assessed using the Log-Rank Test. Other outcome variables included in the study were non-relapse mortality (NRM), which was estimated using the cumulative incidence method, considering graft failure as a competing event. Cumulative incidence of GVHD was calculated, considering death and graft failure as competing events. Univariate analysis using the Cox model with Fine and Gray methods was performed to assess the effect of potential risk factors for GVHD. Multivariate analysis was not performed in this pilot study due to the limited number of patients and low frequency of adverse events. All p-values were 2-sided and, for the statistical analyses, P<0.05 was considered to indicate a statistically significant result. Statistical analysis was performed using version 9.4 of the SAS system for Windows, Copyright© 2002-2012 SAS Institute, Inc., Cary, NC.
Results
Patient and Transplant Characteristics
Baseline characteristics are detailed in Table 1. Median age at transplant was 31 years (range: 18-67) ; 21 (36.2%) recipients were aged over 40 years. The median follow-up of the entire cohort was 6 years (range: 0-13.5). Forty (69%) recipients received grafts from a MSD, 17 (29.3%) from a 10/10 MUD, and 1 (1.7%) case received a graft from a 9/10 MUD. Among the 40 patients who received MSD grafts, 19 (47.5%) had received prior IST. In addition, among the 18 patients who underwent alloHSCT using unrelated donors, 17 (94.5%) had received prior IST. The differences between these proportions were statistically significant (P= 0.007). The source of stem cells was bone marrow (BM) in 46 (79.3%) cases. Alemtuzumab was used as part of the conditioning regimen in 51 (87.9%) recipients and ATG in 7 (12.1%) patients.
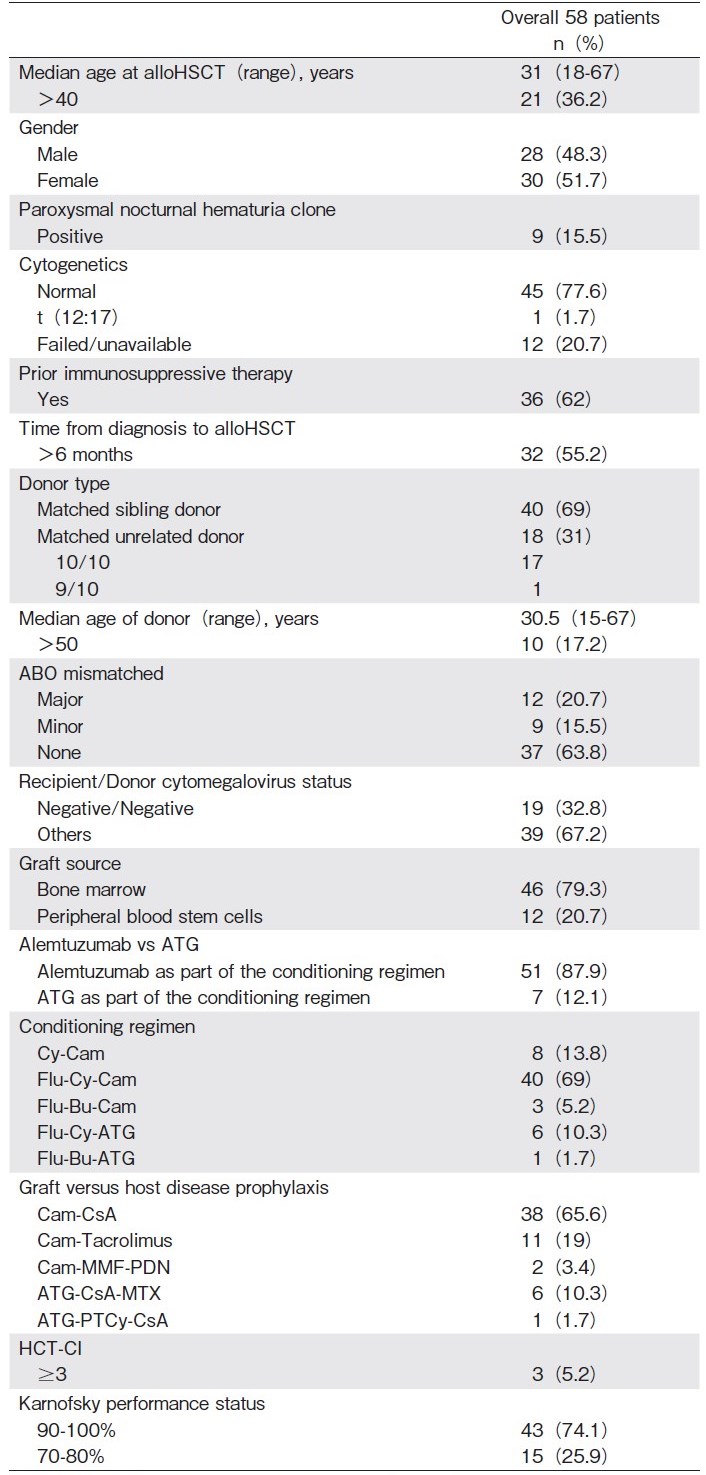
Hematopoietic Recovery and Graft Failure
The median time to neutrophil and platelet engraftment was 18 days (range: 10-100 days) and 17 days (range: 4-100 days), respectively. Among patients who received alemtuzumab, the median time to neutrophil and platelet engraftment was, respectively, 18 days (range: 10-47 days) and 17 days (range: 4-88 days). The median time to engraftment for those recipients who received ATG was, respectively, 15 days (range: 13-24 days) and 23 days (range: 13-44 days). The median time to platelet engraftment was shorter in the cohort of patients who received ATG; these differences were statistically significant (P=0.01). In addition, the times of neutrophil and platelet engraftment were analyzed according to the time of infusion of bone marrow or PBSC grafts. The median time to platelet and neutrophil engraftment was 17.7 and 16 days, respectively, in patients receiving bone marrow grafts, and 15.3 and 14 days, respectively, for patients receiving PBSC grafts. The median time to platelet engraftment was significantly shorter for patients who received BM grafts (P=0.04).
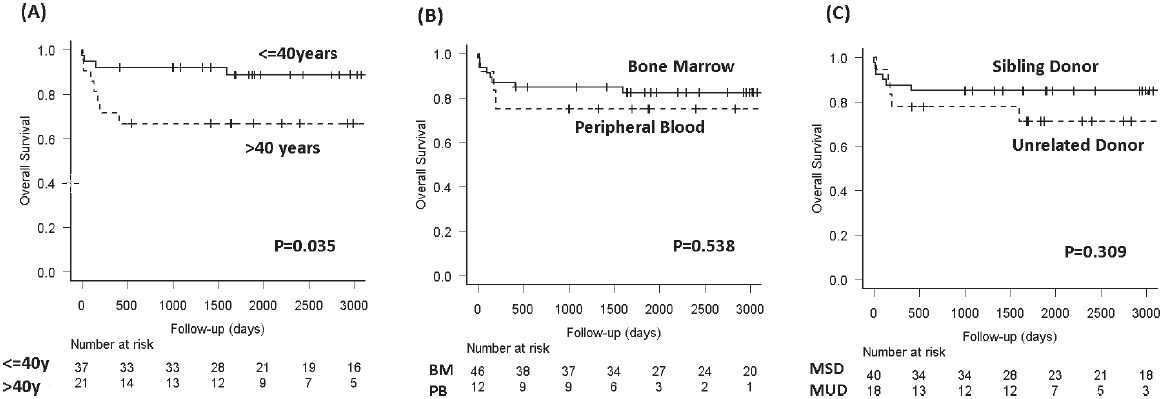
Graft failure (GF) was diagnosed in 8 (13.8%) patients: 3 primary and 5 secondary. All of them had experienced a failure of IST before alloHSCT. One case was successfully treated with CsA, and 5 (62.5%) of the 8 recipients received a second alloHSCT. The other two patients were only eligible for supportive care. Three (37.5%) out of 8 recipients died, secondary to GF. The relationship between GF and relevant baseline characteristics was explored and is summarized in Table 2. The proportion of patients with GF was higher in those who received prior IST (22.2% vs 0%). These differences were statistically significant (P=0.019). In addition, the proportion of patients with GF was comparable for groups of patients older or younger than 40 years, patients with a disease history longer than 6 months? vs shorter than 6 months, donor type (MSD vs MUD), graft source (bone marrow vs peripheral blood) and Karnofsky performance status (KPS) (90-100% KPS vs 70-80% KPS).
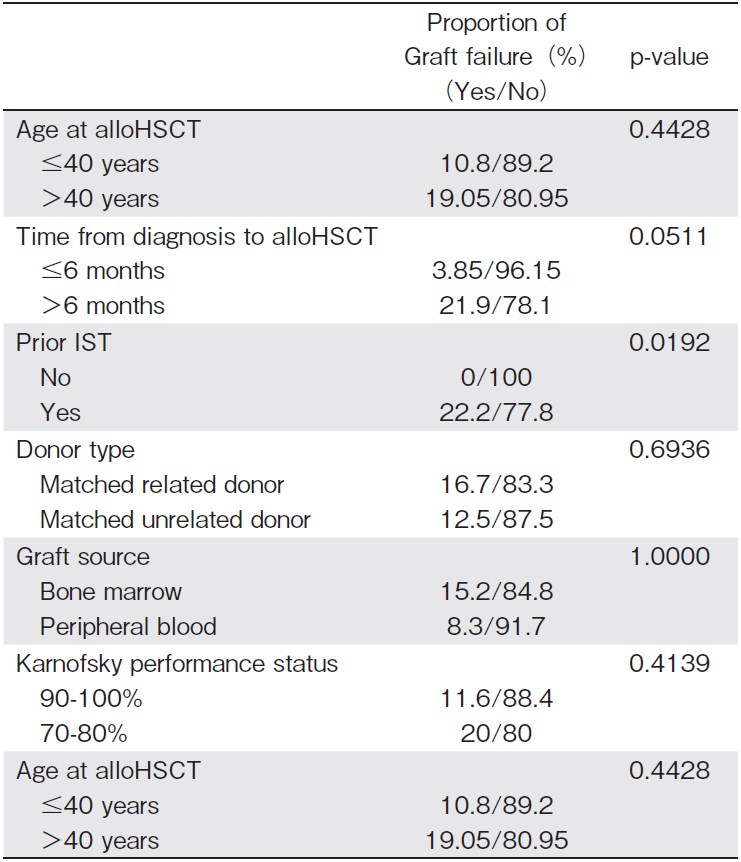
Graft versus Host Disease
The cumulative incidence of GVHD is summarized in the Table 2. Overall, the cumulative incidence of grade ?-? acute GVHD (aGVHD) at day 100 was 13.7% (95% confidence interval (CI) : 4.2-23.1). For patients who received alemtuzumab it was 13.2% (95% CI: 3.4-23.1), and for those who received ATG it was 14.3% (95% CI: 0-40.2). All recipients responded to immuno-suppressive therapy. Age at transplant (?40 years; P=0.466), the use of ATG (P=0.800), donor type (P=0.569), and source of graft (P=0.482) were not significant risk factors to present clinically relevan (t grade ?-?) aGVHD.
The cumulative incidence of any grade of chronic GVHD (cGVHD) at 1 year was 6.9% (95% CI: 0-15.2%). The cumulative incidence of cGVHD at 1 year for patients who received alemtuzumab was 7.8% (95% CI: 0-17%) and for those who received ATG was 0%. All patients responded to immunosuppressive treatment. There was significantly lower risk of developing cGVHD in recipients younger than 40 years (P<0.0001) and those who received ATG (P<0.0001). Neither the selection of MUD grafts (P=0.096) nor the infusion of PBSC grafts (P=0.849) were significant risk factors for cGVHD.
Infections
The main information is summarized in the Table 3. Twenty-three (39.6%) patients had cytomegalovirus (CMV) reactivation, with a median time to reactivation of 34 days (range 21-1023 days). Any result above 200 copies of CMV DNA per milliliter of plasma was considered to be a CMV reactivation and pre-emptive treatment with oral valgancyclovir was initiated until clearance of viremia. No patient had CMV disease. Epstein-Barr virus (EBV) reactivation was seen in 13 (22.4%) patients, with a median time to reactivation of 6.5 months (range: 0-30 days). Any result over>600 copies of EBV DNA per milliliter of plasma was considered an EBV reactivation. Pre-emptive treatment with rituximab was not given to any patient. Five (8.6%) patients were diagnosed with post-transplant lymphoproliferative disorder (PTLD). One case was treated with R-CHOP immuno-chemotherapy and 4 recipients received four doses of rituximab. One patient died because of PTLD. Six (10.3%) recipients had BK cystitis, 16 (27.5%) were diagnosed with other viral infections and four (6.8%) with a fungal infection.
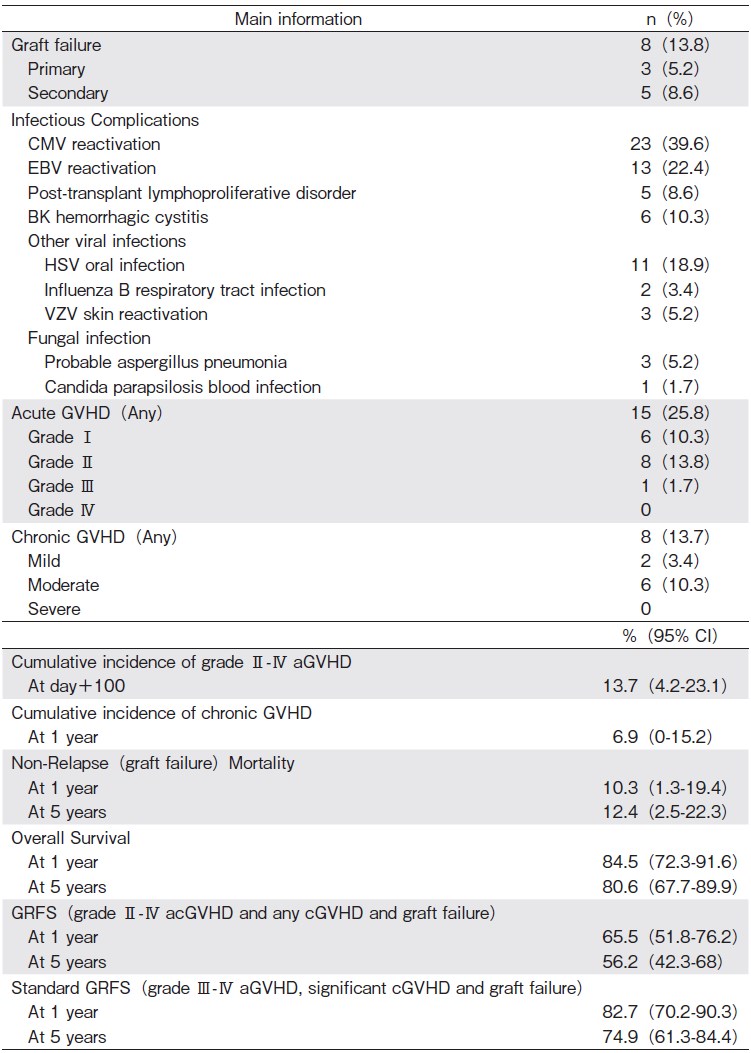
Outcome information
With a median follow-up of 6 years, 11 (18.9%) patients died. Causes of death were as follows: 3 GF, 3 septic shock, 1 PTLD, 1 veno-occlusive syndrome, 2 severe organ failure (1 liver failure and 1 heart failure), and 1 case undocumented.
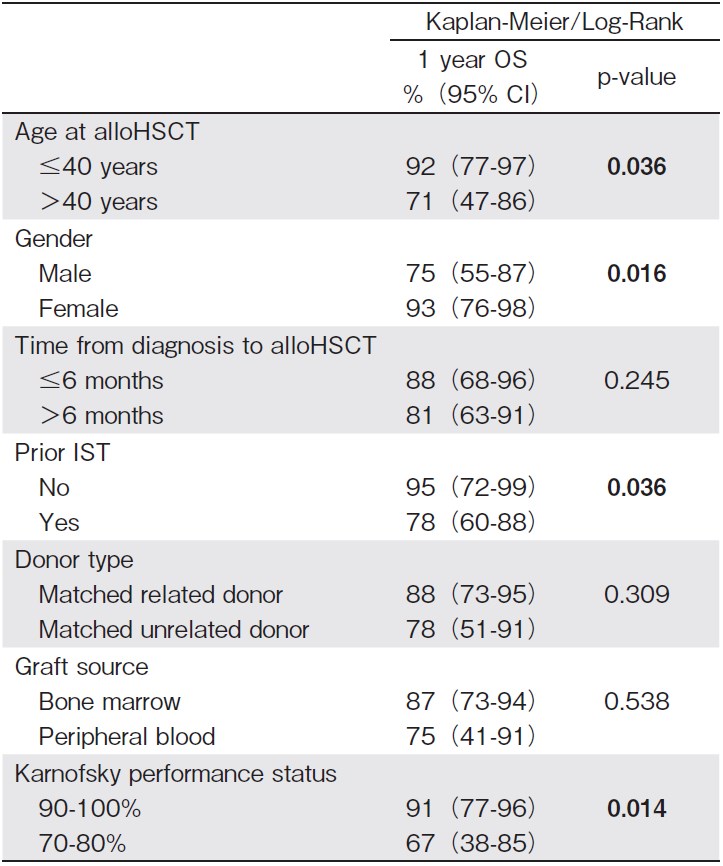
The main outcome information is summarized in the Table 3 and in Figures 1 and 2. One- and five-year OS were, respectively, 84.5% (95% CI: 72.3-91.6) and 80.6% (95% CI: 67.7-89.9). The significant factors for better OS were: age?40 years (P=0.036), female (P=0.016), lack of prior IST (P=0.036), and better performance status prior transplantation (KPS>80%) (P=0.014). Table 4 summarizes the results from the univariate analysis of risk factors for OS.
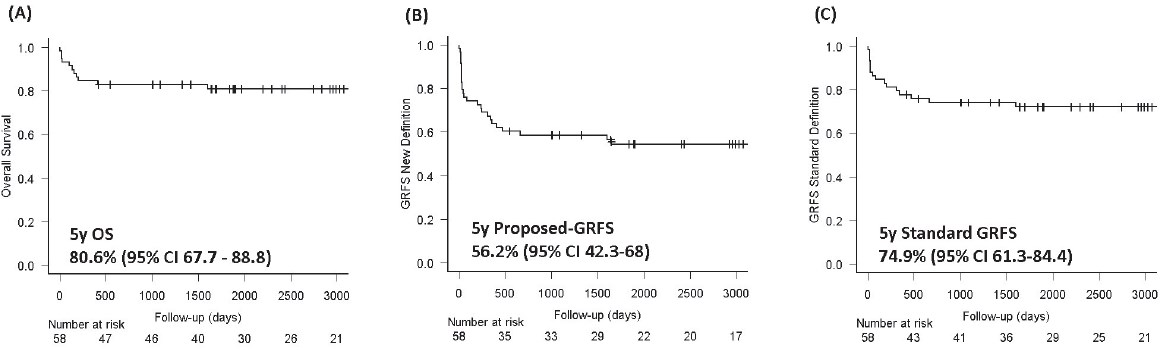
One- and five-year GRFS (calculated to include grade ?-? acute GVHD, any grade of chronic GVHD, graft failure, and death, as events) were, respectively, 65.5% (95% CI: 51.8-76.2) and 56.2% (95% CI: 42.3-68). Oneand five-year GRFS (calculated to include, using the standard definition, grade ?-? acute GVHD, moderate and severe chronic GVHD, graft failure, and death, as events) was 82.7% (95% CI: 70.2-90.3) and 74.9% (9% CI: 61.3-84.4).
Long Term Complications
Among the 47 survivors, eight (17%) recipients developed hypothyroidism and are currently on hormone replacement. One (2.1%) patient developed Graves hyperthyroidism almost 4 years after alloHSCT. One patient had severe autoimmune hemolytic anemia and required a splenectomy. Three (6.3%) recipients developed hemolytic anemia secondary to ABO mismatch. One of these patients was diagnosed with pure red cell aplasia and required up to 6 lines of treatment to control this complication. One patient developed Guillain-Barre syndrome at 9 months after alloHCT and was treated with intravenous immunoglobulins, with good response, and one patient developed "acquired factor ? inhibitor", 13 months post transplantation. One long-term survivor was diagnosed with a smoldering myeloma. This patient had been previously treated with immunosuppressive medication. There were no other cases of malignancy.
Discussion
AlloHSCT upfront, or after failing IST, is used as a curative strategy for patients with SAA. This study details a single-center experience of alloHSCT for SAA, using in vivo T-cell depletion as part of the conditioning regimen, over a time span of 14 years, and reviews the long-term complications.
The overall survival reported in the study is comparable with published data from other single and multicenter studies with a larger number of patients21-23. Several publications support the addition of T-cell depletion as part of the conditioning regimen in SAA, with the aim of reducing graft rejection as well as GVHD rates16-19. The selection of alemtuzumab or ATG depends mainly on center preference13-19.
The term "GRFS" is used as a composite endpoint, focused on survival without relapse and the absence of clinically relevant GVHD, and is primarily used for malignant disorders where there is a benefit from the graft-versus leukemia (GVL) effect24. A novel definition for GRFS, for aplastic anemia, was recently suggested to use rejection-free survival instead of relapse-free survival, maintaining the standard inclusions of grade ?-? cGVHD and clinically significant cGVHD25. We proposed a modification of the original definition of GRFS to include grade ? aGVHD and mild cGVHD in nonmalignant hematological disorders, as there is no benefit from the GVL effect26. Using this definition, the GRFS at 5 years was 56% in our cohort of patients. However, if we use the standard definition of GRFS, it was 74.9%. Using the standard definition, we may overestimate the GRFS in non-malignant hematological disorders, a surrogate marker of quality of life.
Low rates of GVHD were documented in our study. Those recipients who received the ATG-based conditioning regimen had a non-significant trend to lower aGVHD and a significant lower rate of cGVHD. However, the interpretation of these results is limited by the small sample size of the ATG-based cohort and the retrospective design of the study. ATG preserves the expansion of regulatory T-cells, inducing tolerance and controlling alloreactive donor lymphocytes involved in GVHD27,28. In addition, the use of ATG results in a faster achievement of donor chimerism using RIC regimens or alternative donor grafts29,30. Regarding the potential benefit of ATG in engraftment and GVHD control, the use of this drug in SAA patients who undergo alloHSCT is supported by recent publications31.
The use of bone marrow grafts did not have a significant impact on OS and GVHD rates. These results differ from data reported from larger studies32,33. It is generally accepted that BM is the preferred stem cell source in SAA32,33. The use of PBSC grafts reduces the time to engraftment and rates of infection; however, it increases the rate of GVHD34. Among 58 patients who underwent transplant, 4 had an active infection at the time of admission. The presence of an active infection is not a contraindication for alloHSCT in patients with SAA, as the only way to overcome the infection is by neutrophil recovery, which can be achieved by alloHSCT. In infected patients who are critically ill, PBSC may offer earlier myeloid recovery and faster control of infection. Moreover, a donor may not be willing to donate bone marrow, which makes PBSC the only alternative. With more potent T-cell depletion, the adverse effects of PBSC may be partly overcome. Considering recent improvements to the prophylactic approach to GVHD, with the use of ATG or PTCy and the beneficial effects provided by PBSC grafts, the use of this donor source can be considered to reduce time to engraftment in selected patients with SAA and active infections32-36.
Patients aged over 40 years had a higher mortality rate in our study. Despite the improvement of conditioning regimens, GVHD prophylaxis, and supportive care, there remains a higher mortality rate in older recipients diagnosed with SAA3-7. In the setting of SAA, the correlation between age and worse outcome could be related to the fact that older patients are more likely to have had IST before alloHSCT, a longer interval of time between diagnosis and transplant, or worse Karnofsky performance status (KPS) 37,38. In our study, those patients who received prior IST and who had a KPS of 70-80% had a worse post-transplant outcome. Female patients had a better outcome in our cohort. However, the percentage of females prior-treated with IST was lower and the time between diagnosis and alloHSCT was less in this group, than for the male group.
Our study did not find significant superiority in OS for MSD alloHCT compared with MUD alloHCT, possibly due to the limited sample size. The baseline characteristics between patients who received MRD and MUD grafts were well balanced. These results are comparable to studies conducted with a larger number of patients8,9. Again, conclusions from our study are limited by the sample size. However, the lack of significance between MSD and MUD alloHSCT could be due to the refinement of GVHD prophylaxis with the use of T-cell depletion, with either alemtuzumab or ATG. According to international guidelines for management of SAA1-4, patients without a suitable MRD should receive prior immunosuppressive treatment. This increases the time from diagnosis to transplant, the number of transfusions, and infections in this group of patients8,9. With the continuous refinement of GVHD prophylaxis and a better understanding of the physiopathology of GVHD, the upfront indication of alloHSCT, with the selection of MUD or an alternative donor source, could be considered to decrease other well-defined risk factors21,23,39 . At present, AlloHSCT is the only curative strategy for patients with SAA23. In addition, it reduces the risk of clonal evolution to hematological malignancies such as acute myeloid leukemia or myelodysplastic syndromes that occur in about 15% of patients treated with immunosuppressive therapy alone40. The selection of haploidentical donors in SAA is increasing, and early results have been encouraging in both children and adults41-43. Prospective and retrospective studies are needed to compare outcomes between alternative donor sources and matched donors in SAA.
Complications experienced by our patients were primarily immune related, and were seen in 29.6% of patients. The only malignant complication was a smouldering myeloma. This is in contrast to complications seen in transplants performed predominantly for hematological malignancies, where the cumulative incidence of solid, subsequent neoplasms is around 15.2% at 25 years after autologous or allogeneic HSCT44,45. In aplastic anemia, there is a paucity of literature on this aspect8,9. Our study shows that autoimmune complications are predominant. As aplastic anemia is presumed to be an immunemediated disease and the conditioning is non-myeloablative, it may predispose patients to different immunemediated effects, although larger studies are required to determine this.
The main limitations of the study are the heterogeneity of the population included in the analysis, the retrospective design and the small sample size. No multivariate analysis can be done to confirm results obtained in the univariate model.
In conclusion, alloHSCT is a curative strategy for patients with SAA. With the refinement of conditioning regimens, GVHD prophylaxis, and supportive care, the indication of alloHSCT using matched, unrelated donors or alternative donor sources could be considered upfront in selected patients with SAA who do not have a suitable, matched, related donor. Older patients remain a group with higher mortality and more research is needed to improve their outcomes. Long-term complications are mainly autoimmune in nature.
Author Contributions
QS and RK designed the study, interpreted the data and wrote the manuscript. QS collected the data. EA provided the statistical analysis. WL, AL, DK, AG, JL, FM, ZA, AV, JM, VG, and RK provided valuable input into the study design, analysis, and interpretation and reviewed and approved the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.Disclosure forms provided by the authors are available here.
References
1. Killick SB, Bown N, Cavenagh J, Dokal I, Foukaneli T, Hill A, et al. Guidelines for the diagnosis and management of adult aplastic anaemia. Br J Haematol. 2016; 172: 187-207.
2. Bacigalupo A, Giammarco S, Sica S. Bone marrow transplantation versus immunosuppressive therapy in patients with acquired severe aplastic anemia. Int J Hematol. 2016; 104: 168-74.
3. Giammarco S, Peffault de Latour R, Sica S, Dufour C, Socie G, Passweg J, et al. Transplant outcome for patients with acquired aplastic anemia over the age of 40: has the outcome improved? Blood. 2018; 131: 1989-92.
4. Shin SH, Jeon YW, Yoon JH, Yahng SA, Lee SE, Cho BS, et al. Comparable outcomes between younger (40 years) and older (>40 years) adult patients with severe aplastic anemia after HLA-matched sibling stem cell transplantation using fludarabine-based conditioning. Bone Marrow Transplant. 2016; 51: 1456-63.
5. Bacigalupo A, Marsh JC. Unrelated donor search and unrelated donor transplantation in the adult aplastic anaemia patient aged 18-40 years without an HLA-identical sibling and failing immunosuppression. Bone Marrow Transplant. 2013; 48: 198-200.
6. Aljurf M, Al-Zahrani H, Van Lint MT, Passweg JR. Standard treatment of acquired SAA in adult patients 18-40 years old with an HLA-identical sibling donor. Bone Marrow Transplant. 2013; 48: 178-9.
7. Gupta V, Eapen M, Brazauskas R, Carreras J, Aljurf M, Gale RP, et al. Impact of age on outcomes after bone marrow transplantation for acquired aplastic anemia using HLA-matched sibling donors. Haematologica. 2010; 95: 2119-25.
8. Bacigalupo A, Socie G, Hamladji RM, Aljurf M, Maschan A, Kyrcz-Krzemien S, et al. Current outcome of HLA identical sibling versus unrelated donor transplants in severe aplastic anemia: an EBMT analysis. Haematologica. 2015; 100: 696-702.
9. Devillier R, Dalle JH, Kulasekararaj A, D?Aveni M, Clement L, Chybicka A, et al. Unrelated alternative donor transplantation for severe acquired aplastic anemia: a study from the French Society of Bone Marrow Transplantation and Cell Therapies and the EBMT Severe Aplastic Anemia Working Party. Haematologica. 2016; 101: 884-90.
10. Deeg HJ, O?Donnell M, Tolar J, Agarwal R, Harris RE, Feig SA, et al. Optimization of conditioning for marrow transplantation from unrelated donors for patients with aplastic anemia after failure of immunosuppressive therapy. Blood. 2006; 108: 1485-91.
11. Anderlini P, Wu J, Gersten I, Ewell M, Tolar J, Antin JH, et al. Cyclophosphamide conditioning in patients with severe aplastic anaemia given unrelated marrow transplantation: a phase 1-2 dose de-escalation study. Lancet Haematol. 2015; 2: e367-75.
12. Gupta V, Ball SE, Yi QL, Sage D, McCann SR, Lawler M, et al. Favorable effect on acute and chronic graft-versus-host disease with cyclophosphamide and in vivo anti-CD52 monoclonal antibodies for marrow transplantation from HLA-identical sibling donors for acquired aplastic anemia. Biol Blood Marrow Transplant. 2004; 10: 867-76.
13. Gupta V, Ball SE, Sage D, Ortin M, Freires M, Gordon-Smith EC, et al. Marrow transplants from matched unrelated donors for aplastic anaemia using alemtuzumab, fludarabine and cyclophosphamide based conditioning. Bone Marrow Transplant. 2005; 35: 467-71.
14. Hamad N, Del Bel R, Messner HA, Kim D, Kuruvilla J, Lipton JH, et al. Outcomes of hematopoietic cell transplantation in adult patients with acquired aplastic anemia using intermediate-dose alemtuzumab-based conditioning. Biol Blood Marrow Transplant. 2014; 20: 1722-8.
15. Marsh JC, Gupta V, Lim Z, Ho AY, Ireland RM, Hayden J, et al. Alemtuzumab with fludarabine and cyclophosphamide reduces chronic graft-versus-host disease after allogeneic stem cell transplantation for acquired aplastic anemia. Blood. 2011; 118: 2351-7.
16. Kekre N, Zhang Y, Zhang MJ, Carreras J, Ahmed P, Anderlini P, et al. Effect of antithymocyte globulin source on outcomes of bone marrow transplantation for severe aplastic anemia. Haematologica. 2017; 102: 1291-8.
17. Bacigalupo A, Socie G, Lanino E, Prete A, Locatelli F, Locasciulli A, et al. Fludarabine, cyclophosphamide, antithymocyte globulin, with or without low dose total body irradiation, for alternative donor transplants, in acquired severe aplastic anemia: a retrospective study from the EBMT-SAA Working Party. Haematologica. 2010; 95: 976-82.
18. Chen X, Wei J, Huang Y, He Y, Yang D, Zhang R, et al. Effect of Antithymocyte Globulin Source on Outcomes of HLAMatched Sibling Allogeneic Hematopoietic Stem Cell Transplantation for Patients with Severe Aplastic Anemia. Biol Blood Marrow Transplant. 2018; 24: 86-90.
19. Marsh JC, Pearce RM, Koh MB, Lim Z, Pagliuca A, Mufti GJ, et al. Retrospective study of alemtuzumab vs ATG-based conditioning without irradiation for unrelated and matched sibling donor transplants in acquired severe aplastic anemia: a study from the British Society for Blood and Marrow Transplantation. Bone Marrow Transplant. 2014; 49: 42-8.
20. Champlin RE, Perez WS, Passweg JR, Klein JP, Camitta BM, Gluckman E, et al. Bone marrow transplantation for severe aplastic anemia: a randomized controlled study of conditioning regimens. Blood. 2007; 109: 4582-5.
21. Georges GE, Doney K, Storb R. Severe aplastic anemia: allogeneic bone marrow transplantation as first-line treatment. Blood Adv. 2018; 2: 2020-8.
22. Sun Q, Wu B, Zhu Z, Sun C, Xu J, Long H, et al. Allogeneic Hematopoietic Stem Cell Transplant for Severe Aplastic Anemia: Current State and Future Directions. Curr Stem Cell Res Ther. 2018; 13: 350-5.
23. Kumar R, Bonfim C, George B. Hematopoietic cell transplantation for aplastic anemia. Curr Opin Hematol. 2017; 24: 509-14.
24. Holtan SG, DeFor TE, Lazaryan A, Bejanyan N, Arora M, Brunstein CG, et al. Composite end point of graft-versus-host disease-free, relapse-free survival after allogeneic hematopoietic cell transplantation. Blood. 2015; 125: 1333-8.
25. Vaht K, Goransson M, Carlson K, Isaksson C, Lenhoff S, Sandstedt A, et al. High Graft-versus-Host Disease-Free, Relapse/Rejection-Free Survival and Similar Outcome of Related and Unrelated Allogeneic Stem Cell Transplantation for Aplastic Anemia: A Nationwide Swedish Cohort Study. Biol Blood Marrow Transplant. 2019; 25: 1970-4.
26. Salas MQ, Kumar R. Graft-versus-Host Disease-Free Relapse-Free Survival Definition for Patients with Nonmalignant Hematologic Disorders. Biol Blood Marrow Transplant. 2020; 26: 428.
27. Feng X, Kajigaya S, Solomou EE, Keyvanfar K, Xu X, Raghavachari N, et al. Rabbit ATG but not horse ATG promotes expansion of functional CD4+CD25highFOXP3+regulatory T cells in vitro. Blood. 2008; 111: 3675-83.
28. Lopez M, Clarkson MR, Albin M, Sayegh MH, Najafian N. A novel mechanism of action for anti-thymocyte globulin: induction of CD4+CD25+Foxp3+ regulatory T cells. J Am Soc Nephrol. 2006; 17: 2844-53.
29. Bacigalupo A, Lamparelli T, Bruzzi P, Guidi S, Alessandrino PE, di Bartolomeo P, et al. Antithymocyte globulin for graftversus-host disease prophylaxis in transplants from unrelated donors: 2 randomized studies from Gruppo Italiano Trapianti Midollo Osseo (GITMO). Blood. 2001; 98: 2942-7.
30. Walker I, Panzarella T, Couban S, Couture F, Devins G, Elemary M, et al. Pretreatment with anti-thymocyte globulin versus no anti-thymocyte globulin in patients with haematological malignancies undergoing haemopoietic cell transplantation from unrelated donors: a randomised, controlled, openlabel, phase 3, multicentre trial. Lancet Oncol. 2016; 17: 164-73.
31. Bacigalupo A. Antithymocyte globulin and transplants for aplastic anemia. Haematologica. 2017; 102: 1137-8.
32. Bacigalupo A, Socie G, Schrezenmeier H, Tichelli A, Locasciulli A, Fuehrer M, et al. Bone marrow versus peripheral blood as the stem cell source for sibling transplants in acquired aplastic anemia: survival advantage for bone marrow in all age groups. Haematologica. 2012; 97: 1142-8.
33. Kumar R, Kimura F, Ahn KW, Hu ZH, Kuwatsuka Y, Klein JP, et al. Comparing Outcomes with Bone Marrow or Peripheral Blood Stem Cells as Graft Source for Matched Sibling Transplants in Severe Aplastic Anemia across Different Economic Regions. Biol Blood Marrow Transplant. 2016; 22: 932-40.
34. Stamatovic D, Balint B, Tukic L, Elez M, Tarabar O, Todorovic M, et al. Impact of stem cell source on allogeneic stem cell transplantation outcome in hematological malignancies. Vojnosanit Pregl. 2011; 68: 1026-32.
35. Sanz J, Moscardo F, Montoro J, Cano I, Guerreiro M, Dasi MA, et al. Partial T Cell-Depleted Peripheral Blood Stem Cell Transplantation from HLA-Identical Sibling Donors for Patients with Severe Aplastic Anemia. Biol Blood Marrow Transplant. 2020; 26: 83-7.
36. Kim H, Lee KH, Sohn SK, Kim I, Kim SH, Park Y, et al. Effect of Stem Cell Source and Dose on Allogeneic Hematopoietic Stem Cell Transplantation in Adult Patients with Idiopathic Aplastic Anemia: Data from the Korean Aplastic Anemia Trials. Acta Haematol. 2020; 143: 232-43.
37. Georges GE. Consider Allogeneic Bone Marrow Transplantation for Older, FitPatients with Aplastic Anemia. Biol Blood Marrow Transplant. 2019; 25: e69-e70.
38. Rice C, Eikema DJ, Marsh JCW, Knol C, Hebert K, Putter H, et al. Allogeneic Hematopoietic Cell Transplantation in Patients Aged 50Years or Older with Severe Aplastic Anemia. Biol Blood Marrow Transplant. 2019; 25: 488-95.
39. Marsh JCW, Risitano AM, Mufti GJ. The Case for Upfront HLA-Matched Unrelated Donor Hematopoietic Stem Cell Transplantation as a Curative Option for Adult Acquired Severe Aplastic Anemia. Biol Blood Marrow Transplant. 2019; 25: e277-e84.
40. Young NS, Ogawa S. Somatic Mutations and Clonal Hematopoiesis in Aplastic Anemia. N Engl J Med. 2015; 373: 1675-6.
41. Ebens CL, DeFor TE, Tryon R, Wagner JE, MacMillan ML. Comparable Outcomes after HLA-Matched Sibling and Alternative Donor Hematopoietic Cell Transplantation for Children with Fanconi Anemia and Severe Aplastic Anemia. Biol Blood Marrow Transplant. 2018; 24: 765-71.
42. Li Y, Duan F, Xiao H, Wu X, Wang S, Xu D, et al. Therapeutic Outcomes of Haploidentical Allogeneic Hematopoietic Stem Cell Transplantation in Patients With Severe Aplastic Anemia: A Multicenter Study. Transplantation. 2018; 102: 1724-31.
43. DeZern AE, Brodsky RA. Haploidentical Donor Bone Marrow Transplantation for Severe Aplastic Anemia. Hematol Oncol Clin N Am. 2018; 32: 629-42.
44. Bhatia S, Armenian SH, Landier W. How I monitor long-term and late effects after blood or marrow transplantation. Blood. 2017; 130: 1303-14.
45. Inamoto Y, Shah NN, Savani BN, Shaw BE, Abraham AA, Ahmed IA et al. Secondary solid cancer screening following hematopoietic cell transplantation. Bone Marrow Transplantation. 2015; 50: 1013-23.
News


